![]()
![]()
![]()
El valor nutricional de un alimento depende en gran medida de la calidad de los ingredientes utilizados, la cual se mide en función de su capacidad de promover el crecimiento y mantener en buen estado a los organismos. En este sentido, el origen y/o el tratamiento previo a que se someten los materiales influirá sobre su calidad, en función de la disponibilidad de nutrientes para usarse en las funciones metabólicas del animal, o también, el contenido de factores tóxicos endógenos característicos de cada material, particularmente aquellos de origen vegetal, que funcionan como antinutrientes afectando negativamente al organismo.
En este sentido, existen varias técnicas que permiten conocer la disponibilidad de un nutriente determinado, principalmente aminoácidos, así como también, determinar el contenido de sustancias tóxicas a fin de eliminarlas o desechar el material. En este documento se incluyen los métodos para análisis de los antinutrientes más comunes encontrados en las proteínas vegetales.
Este método determina la ureasa residual en harina de soya y sus derivados de acuerdo al método Caskey-Knapp (1944) modificado por AACC (1969) y Quím. Leticia Comar (Nutrimentos del Sureste, Mérida, Yucatán, México). El resultado esta dado en unidades de pH proporcionales a la actividad ureásica. Los valores aceptables oscilan entre 0.05 y 0.5; valores menores indican sobrecocimiento y mayores falta de cocimiento.
Reactivos
Materiales y Equipo
Procedimiento
Pese 0.2g de harina de soya finamente molida (sin sobrecalentar) por duplicado y colocar en tubos de ensaye (A y B); adicione al tubo A 10ml de la solución bufer de urea. Tape, agite e incube en baño maría por 30 min a 30 ± 0.5°C.
Después de 5 min, adicione al tubo B 10ml de sol. bufer de fosfatos, tape, agite y coloque en el baño maría.
Agitar cada uno de los tubos cada 5 minutos (seis agitaciones en 30 min).
Retire el tubo A del baño maría, decante el sobrenadante en un vaso de pp de 10ml y mida el pH dentro de un período menor a 3 min. Repita el procedimiento con el tubo B 5 minutos después del A.
Calcule el índice de actividad ureásica como unidades de pH resultantes de la diferencia de las lecturas del tubo A menos el tubo B y determine la calidad de la muestra según la tabla de ejemplo incluida:
IAU= pH tubo A (muestra + bufer urea) - pH tubo B (muestra + bufer fosfatos)
| Grado de cocimiento | Unidades de pH | pH tubo A | pH tubo B | Color |
| Cruda | 1.9 | 8.80 | 6.90 | Rojo |
| Subcocida | 0.80 | 7.60 | 6.80 | Rosa |
| Cocido adecuado | 0.30 | 7.10 | 6.80 | Rosa |
| Cocido adecuado | 0.08 | 6.98 | 6.90 | Rosa tenue |
| Sobrecocida | 0.03 | 6.93 | 6.90 | Ambar |
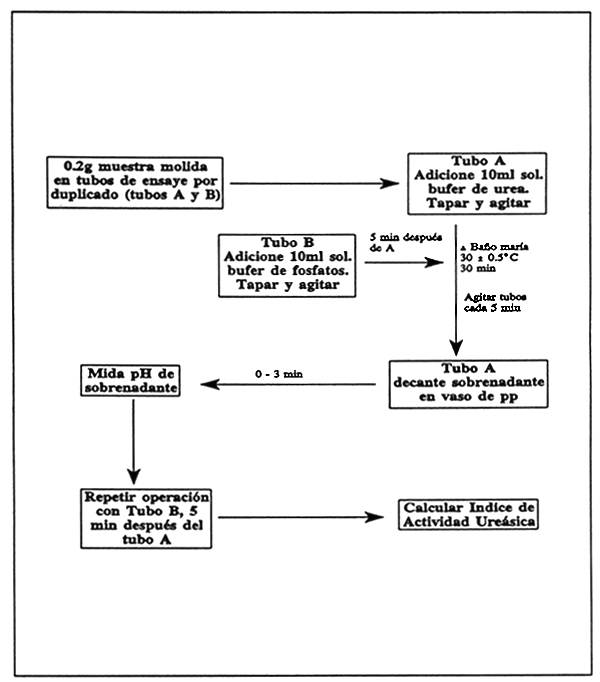
FIGURA 24
Determinación de actividad ureásica en harina de soya y sus derivados.
Este antinutriente es característico de la harina de algodón, siendo necesario determinar su contenido debido a sus efectos adversos sobre los organismos alimentados con la semilla, aun cuando los peces aparentemente resisten mayores concentraciones del alcaloide debido al elevado contenido proteico de su dieta. En este documento se describen dos métodos, el primero para harinas normales y el segundo para harinas que han sido tratadas químicamente y contienen dianilinogosipol.
Reactivos:
Disuelva 25 mg de gosipol puro en acetona libre de anilina y transfiera a un matraz aforado de 250 ml, usando 100 ml de acetona. Adicione 75 ml de agua destilada, afore con acetona y mezcle.
Tome 50 ml de la solución (a), adicione 100 ml de acetona pura, 60 ml de agua destilada, mezcle y diluya a 250 ml con acetona pura. La solución (b) contiene 0.02 mg de gosipol/ml y es estable por 24 h en oscuridad.
Materiales y Equipo
Procedimiento
Muela la muestra para que pase la malla de 1 mm, cuidando de no sobrecalentarla. Tome aproximadamente 1 g de la muestra y agréguele 25 ml de acetona pura. Agite por unos minutos, filtre y divida el filtrado en dos. A una porción, adiciónele una lenteja de Hidróxido de Sodio y caliente en baño maría por unos minutos. Un extracto ligeramente amarillo que no cambia de color con el Hidróxido indica que la harina no ha sido tratada, proceda con el método 1. Si se obtiene un color rojo-anaranjado profundo, indica la presencia de dianilinogosispol y requiere del método 2.
Método 1
Pese 0.5–1 g de muestra, dependiendo de la cantidad de gosipol esperada, en un matraz erlenmayer y adicione 2 ó 3 perlas de vidrio. Agregue 50 ml de solución de acetona acuosa, tape el matraz y agite por una hora. Filtre, deseche los primeros mililitros de filtrado y luego tome dos alícuotas iguales (2–10 ml, dependiendo del contenido de gosipol esperado) y páselas a matraces volumétricos de 25 ml. Diluya una de las alícuotas, afore a volumen con alcohol isopropílico acuoso (solución a), mientras la otra alícuota (solución b). Adiciónele 2 ml de anilina purificada, caliente en baño maría (90–100°C) por 30 min. junto con un blanco conteniendo 2 ml de anilina y un volumen de solución acuosa de acetona igual a la alícuota. Retire la solución b y el blanco, adicione suficiente alcohol isopropílico acuoso para efectuar solución homogénea, y enfría a temperatura ambiente en un baño con agua. Afore con alcohol isopropílico acuoso.
Lea las muestras a una absorvancia de 400 nm en el espectrofotómetro. Ajuste el instrumento a 0 absorvancia con alcohol isopropílico acuoso y determine la absorvancia de la solución (a) y el blanco de reactivo. Si el blanco está abajo de 0.022 de absorvancia continúe con el análisis, de lo contrario repita el análisis usando anilina recién destilada.
Determine la absorvancia de la solución (b) con el blanco de reactivo ajuste a 0 absorvancia. Calcule la absorvancia corregida de la alícuota: absorvancia de la solución (b) menos absorvancia de la solución (a). Determine los mg de gosipol libre presentes en la solución de la muestra usando la curva de calibración.
Método 2.
Pese 1 g de muestra en un matraz erlenmayer, agregue 50 ml de acetona acuosa, agite y filtre como el anterior. Agregue alícuotas duplicadas del filtrado (de 2 a 5 ml, dependiendo de la cantidad esperada de gosipol libre) a matraces volumétricos de 25 ml. Afore una de las alícuotas (solución a) con el alcohol isopropílico acuoso y déjelo reposar 30 minutos antes de leerlo en el espectrofotómetro. Afore la otra alícuota (solución b) como en el procedimiento (1), determine las absorvancias de las soluciones a y b como el procedimiento anterior y calcule el contenido aparente de gosipol de ambas soluciones usando la curva de calibración.
Preparación de la Curva de Calibración.
Tome alícuotas duplicadas de 1, 2, 3, 4, 5, 7, 8 y 10 ml de estándar de gosipol 0.02 mg/ml y colóquelas en matraces volumétricos de 25 ml. Diluya una serie (solución A) aforando con alcohol isopropílico acuoso y determine la absorvancia como en el anterior. A la otra serie (solución b) adiciónele 2 ml de anilina purificada y proceda como anteriormente. Prepare un blanco de reactivos usando 2 ml de anilina y 10 ml de acetona acuosa, calentada junto con el estándar. Determine absorvancias como en el procedimiento (1) y calcule la densidad óptica corregida para cada solución estándar:
Absorvancia Corregida = (Abs. solución b - Abs. solución a)
Trace la curva estándar, graficando absorvancia corregida contra concentración de gosipol en 25 ml.
Calule % de gosipol libre en harinas normales.
Gosipol libre % = 5G/WV
Donde:
G= Lectura en la Gráfica
W= Peso de la muestra
V= Volumen de la alícuota usada.
Para harinas químicamente tratadas.
Gosipol libre % = 5(B - A)/WV
Donde:
A = mg Gosipol libre aparente en alícuota (a)
B= mg Gosipol libre aparente en alícuota (b)
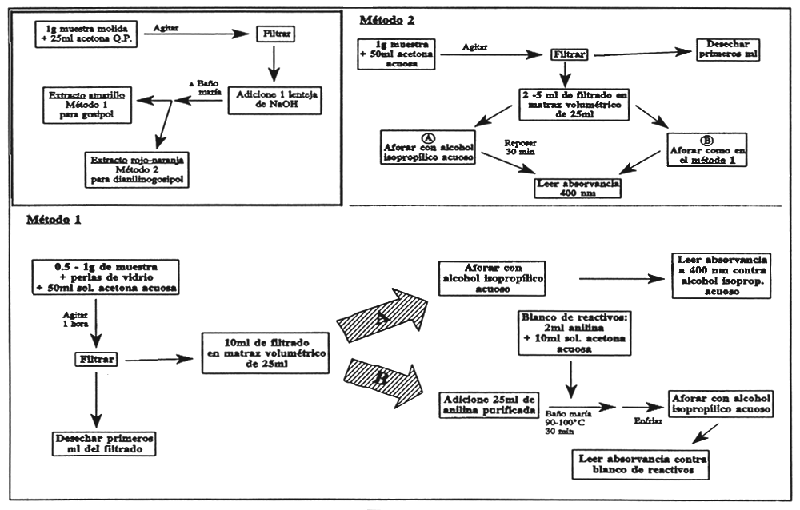
FIGURA 25
Determinación de gosipol libre en harinas de semilla de algodón.
El método descrito proporciona el contenido aproximado de tioglucósidos pero no determina tioglucósidos e isocianatos individuales.
Reactivos y Aparatos
Procedimiento
A 10 g de harina (desengrasada por extracción Soxhlet) adiciónele 250 ml de agua destilada, hidrolice a 54°C por 1 h y caliente a ebullición por 2 h., manteniendo el volumen constante.
Filtre, reteniendo el filtrado y lave el residuo tres veces con 50 ml de agua caliente, agregando el líquido al filtrado inicial y afore a 600 ml.
Precipite el sulfato de bario por calentamiento y adicionando un exceso de solución de Cloruro de bario. Déjelo en un baño de vapor por algunas horas y filtre.
Calcine en una mufla y pese el precipitado.
Calcule el contenido aproximado de tioglucósido como:
% Tiogl= (M. wt. tioglucósido × Peso de BaSO4) × 100/M.wt. BaSO4 × Peso Muestra
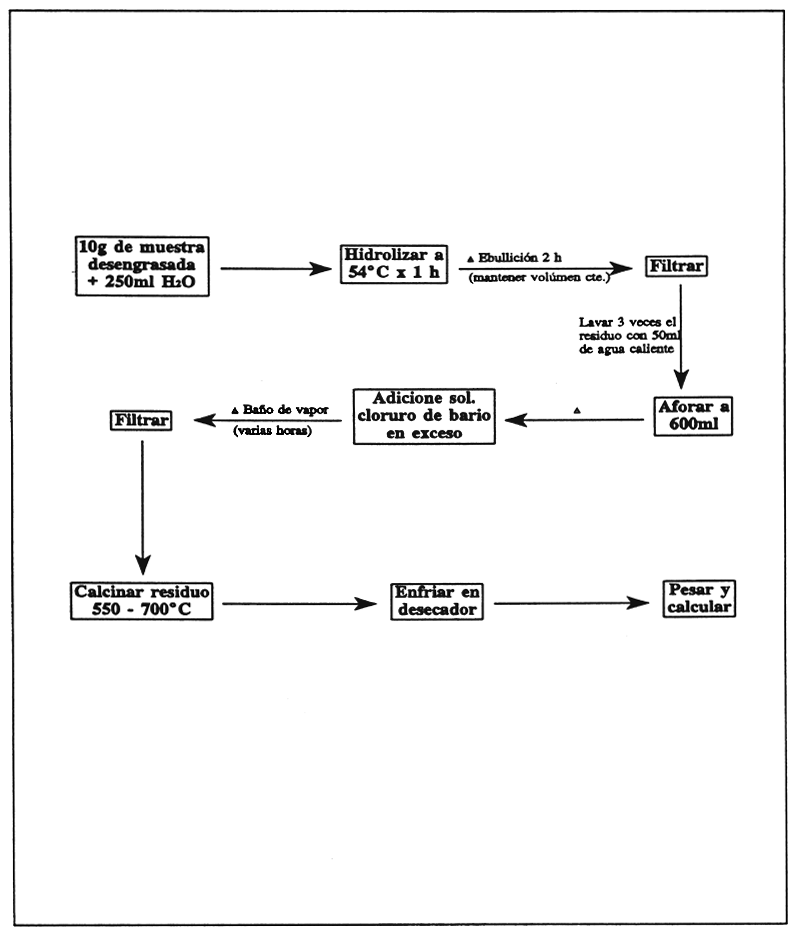
FIGURA 26
Determinación de tioglucósidos en ingredientes alimenticios.
El método que se incluye es adecuado para materiales como harina de cacahuate, harina de copra y harina de palma kernel; para detalles del método, así como para procedimientos alternativos, refiérase a Methods of Aflatoxin Analysis por B.D. Jones (1972), Report No. G70, Tropical Products Institute, London.
Reactivos:
Materiales y Equipo
Procedimiento
Pese 10 mg del material en un frasco de boca ancha y mézclelo con 10 ml de agua (si se usa material con alto nivel de grasa, será necesaria una extracción previa con soxhlet). Adicione 100 ml de cloroformo y agite por 30 min.
Filtre el extracto a través de la celita, tome 20 ml del filtrado y llévelo a 25 ml (solución a). Tome otros 20 ml del filtrado y concéntrelos a 5 ml (solución b).
Prepare placas de cromatografía agitando 100 g de gel de sílica “G” con 220 ml de agua por 20 min y aplicando la mezcla a las placas con el aparato apropiado, con un grosor de 509μ. Déjela reposar por 1 hora y seque a 100°C.
Coloque gotas de 10 y 20 μl de solución b y 15 y 10μl de solución a en la placa, junto con una gota de estándar cualitativo, en una línea a 2 cm del fondo de la placa y al menos a 2 cm de cada lado. Realice la aplicación bajo luz tenue.
Revele la placa en éter etílico a una altura de 12 cm, déjela secar en luz tenue y luego vuélvala a revelar en mezcla de cloroformo/metanol hasta una altura de 10 cm desde la línea basal.
Examine la placa en un cuarto obscuro a 30 cm de la fuente de UV. La presencia de una mancha azul fluorescente a Rf 0.5–0.55 indica aflatoxina B (asegúrese de que la mancha del estándar se encuentre también en ese rango). la presencia de una segunda mancha a Rf 0.45 – 0.5 indica aflatoxina G.
La toxicidad de una muestra puede ser clasificada en términos de aflatoxinas B y G de acuerdo con la tabla siguiente:
| Volumen aplicado (ml) | Conc. de Aflatoxina (mg/kg) | Toxicidad de fluorescencia observada | |
| No fluoresencia | c/fluoresencia | ||
| 5 (sol. A) | <1000 | >1000 | Muy alta |
| 10 (sol. A) | <500 | 500–1000 | Alta |
| 10 (sol. B) | <100 | 100–500 | Media |
| 20 (sol. B) | <50 | 50–100 | Baja |
Tomado de Harris, 1980
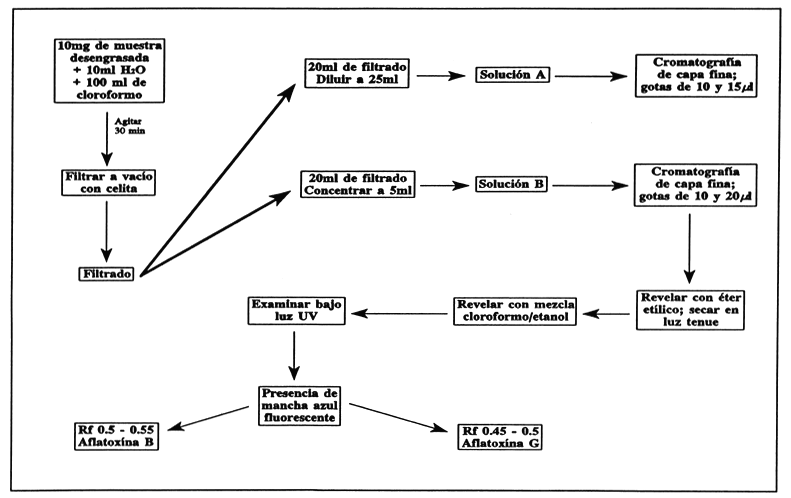
FIGURA 27
Determinación cromatográfica de aflatoxinas en ingredientes alimenticios.
La mimosina es un aminoácido libre muy común en algunas leguminosas, incluyendo a Leucaena leucocephala y L. glauca, consideradas excelentes fuentes de proteína para la alimentación animal. Su presencia limita el uso de las hojas y semillas en la alimentación de monogástricos, ya que afecta la función de la tiroides, provocando reducción del crecimiento.
Reactivos
Materiales y Equipo
Procedimiento
Extracción: Coloque 1.25 g de hoja seca de leucaena en un Vaso de precipitado de 250 ml y digiera con 100 ml de HCl 0.1N por hora con agitación constante, deje enfriar y transfiera todo a un matraz volumétrico de 250 ml, agite bien y deje reposar hasta que los sólidos se asienten en el fondo.
Clarificación: Transfiera 10 ml de sobrenadante a un V.P. de 150 ml con 30 mg de Carbón. Agregue agua para llevar el volumen a 25 ml, cúbralo con un vidrio de reloj y caliente a ebullición por 15 min., deje enfriar y filtre con vacío en el crisol de filtración; el filtrado es colectado en un tubo colocado en el matraz kitazato. Enjuague el vaso de precipitado y el material en el crisol con 10 ml de HCl 0.1N dividido en tres porciones. Enjuague finalmente el crisol con 5 pequeñas porciones de agua.
Determinación Colorimétrica:
Transfiera el líquido obtenido a un matraz volumétrico de 100 ml, agregue 4 ml de solución de cloruro férrico al 0.5% en HCl 0.1N y afore con agua. El pH deberá ser entre 1.5 y 2.5. Lea en el Colorímetro y calcule la cantidad de mimosina en una curva de calibración.
Curva de Calibración: Disuelva 0.1046 g de mimosina pura en HCl 0.1N y afore a 100 ml con el ácido. La solución contiene 0.1% de mimosina. Coloque 1 ml de solución de mimosina en un matraz volumétrico de 100 ml y adicione 10 ml de HCl 0.1N y 4 ml de la solución de cloruro férrico al 0.5% y afore con agua. Lea la intensidad del color en el Colorímetro (espectro 535 nm). Use agua destilada como referencia y repita el procedimiento usando 2,3,4 y 5 ml de solución de mimosina. Se obtiene una línea al graficar la lectura del Colorímetro contra la concentración.
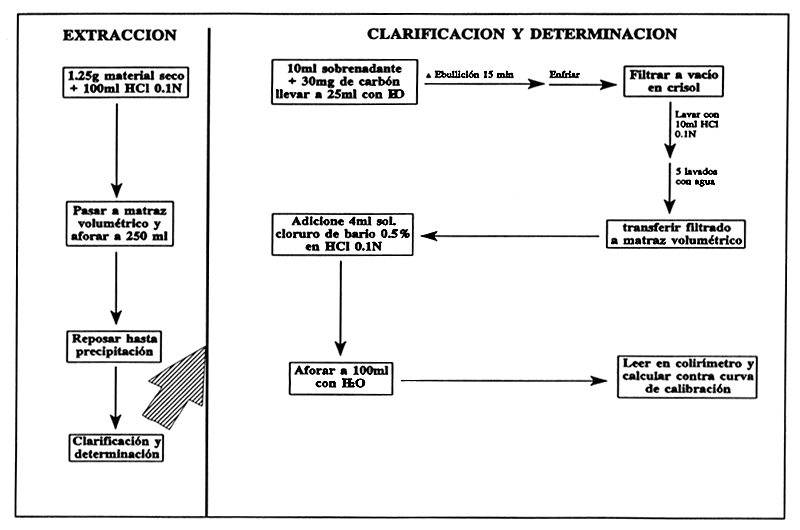
FIGURA 28
Determinación colorimétrica de mimosina.
Las semillas y hojas de leguminosas y en particular las de canavalia (Canavalia ensiformis) y sesbania (Sesbania spp.) contienen cantidades variables del aminoácido libre canavanina, el cual es tóxico para animales monogástricos. El método permite determinar la cantidad del antinutriente en ingredientes alimenticios.
Reactivos
Materiales y Equipo
Procedimiento
Ponga en un tubo de ensaye una muestra de 1 ml de extracto de la muestra, conteniendo 0–0.25 mg de canavanina, se le adicionan 0.5 ml de buffer fosfato y 0.5 ml de reactivo de acuoprusiato. Maneje de la misma manera blancos conteniendo 0.2, 0.5, 0.8, y 1 ml de solución madre diluida y llevados a 1 ml con agua, así como también un blanco de reactivos con 1 ml de agua, tratados con el fosfato y acuoprusiato.
Mezcle vigorosamente y guarde los tubos en un gabinete obscuro; se desarrollará una coloración rojiza en presencia de canavanina.
Después de 2 horas lea en un colorímetro o espectrofotómetro. En este último, ajuste a una longitud de onda de 520 nm y el blanco a una densidad óptica de cero. Puede usar cubetas del espectrofotómetro para desarrollar el color; si usa cubetas de más de 2 ml, puede adicionarles hasta 10 ml de agua después de las 2 horas de incubación para desarrollo del color.
Cálculos
Se grafica la densidad óptica de cada estándar contra su cantidad correspondiente de canavanina y el contenido de canavanina de las muestras se calcula a partir de la curva.
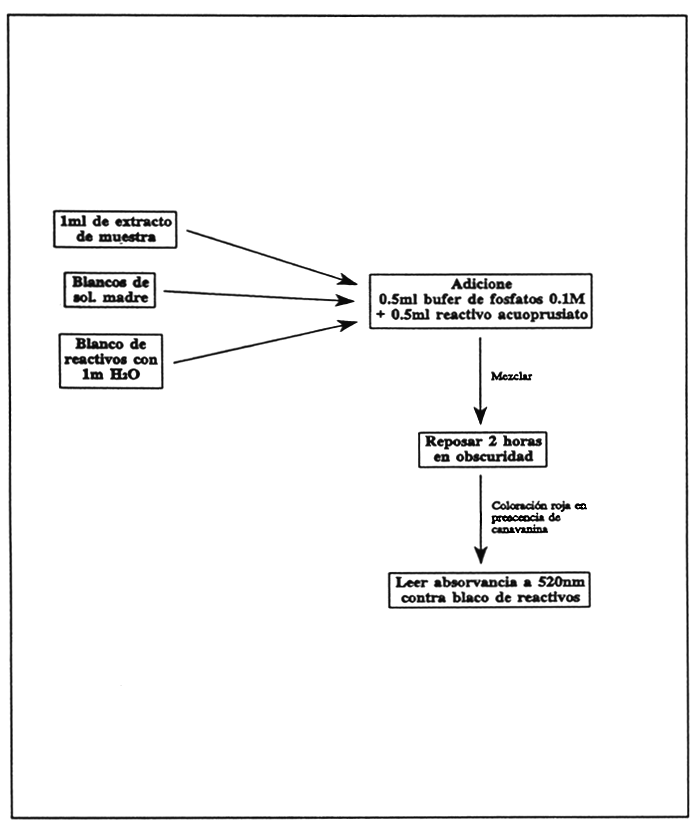
FIGURA 29
Determinación colorimétrica de canavanina en ingredientes alimenticios.
El ácido cianhídrico se encuentra de manera natural en diferentes materiales como son la semilla de lino, harina de yuca y algunas semillas de leguminosas. Este método permite determinar el ácido libre o en forma de glucósido, para lo cual, la muestra se suspende en agua, se libera el ácido mediante enzimas y se separa por destilación para colectarse en una solución de nitrato de plata acidificada.
Reactivos
Materiales y Equipo
Procedimiento
Pese con exactitud de 0.005g aproximadamente 20g de la muestra preparada, colóquela en un matraz de fondo plano de 1000 ml, adicione 50 ml de agua y 10 ml de suspensión de almendras dulces. Tape el matraz y póngalo en la estufa durante 16 horas a 37–38°C. Enfríe a temperatura ambiente, adicione 80 ml de agua, 10 ml de solución de acetato de sodio y una gota de antiespumante.
Conecte el matraz al aparato de destilación y colóquelo en el baño de aceite previamente ajustado a una temperatura ligeramente superior a 100°C; destile 200–300 ml de líquido pasando una corriente de vapor a través del matraz y calentando suavemente el baño de vapor. Colecte el destilado en un matraz erlenmayer protegido de la luz conteniendo exactamente 50ml de solución de nitrato de plata 0.02N y 1ml de ácido nítrico. Asegúrese de que la extensión del condensador esté sumergida en la solución de nitrato de plata.
Transfiera el contenido del matraz erlenmayer a un matraz aforado de 500 ml y llévelo al volumen con agua, mezcle y filtre. Remueva 250 ml del filtrado, adicione aproximadamente 1 ml de sulfato férrico amoniacal y titule el exceso de nitrato de plata con la solución de tiocianato de amonio. Si se desea, se puede correr un blanco siguiendo el mismo procedimiento con 10ml de suspensión de almendras dulces omitiendo la muestra.
Expresión de resultados
Si el blanco indica que la solución de nitrato de plata ha sido consumida, reste su valor del volumen consumido por la destilación de la muestra. 1 ml de 0.02N AgNO3 º 0.54mg de HCN. Exprese el resultado como porcentaje de la muestra.
NOTA: Si la muestra (ej. frijoles) contiene una elevada cantidad de azufre, se formará un precipitado negro de sulfato de plata, el cual se filtra junto con el depósito de cianuro de plata. La formación de este precipitado consume solución de nitrato de plata y su efecto debe ser eliminado para el cálculo de contenido de HCN, para lo cual, trate el depósito sobre el papel filtro con 50ml de amoníaco a fin de disolver el cianuro de plata. Lave el residuo en amoníaco diluido y determine su contenido de plata. Convierta el valor a ml de solución 0.02N de solución de nitrato de plata y résteselo al volumen de solución de nitrato de plata consumida por el destilado de la muestra.
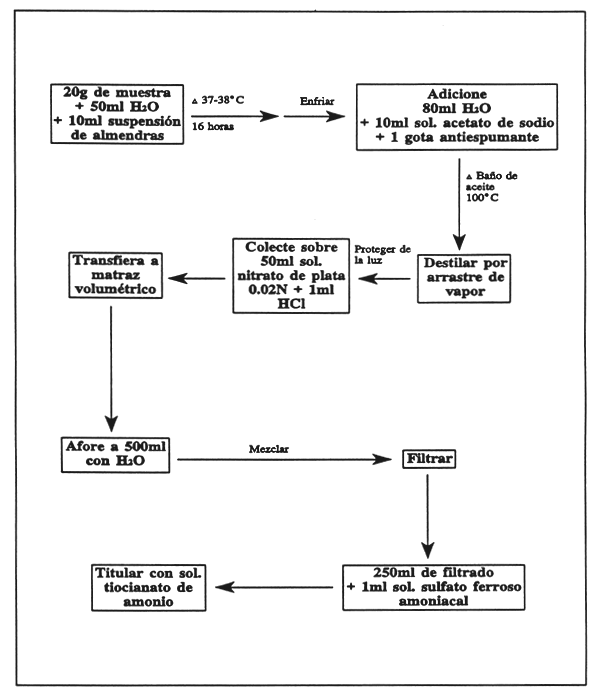
FIGURA 30
Determinación de ácido cianhídrico en ingredientes alimenticios.
Los taninos son glucósidos de polipéptidos solubles en agua presentes en muchas plantas, con un sabor agrio astringente, confiere sabor y olor indeseable a los alimentos. Afectan la utilización de las proteínas debido a que ligan a la lisina, haciéndola indisponible para animales monogástricos.
Reactivos
Procedimiento
Extraiga 2g de muestra durante 20 horas con éter anhidro. Hierva el residuo por 2 horas con 300ml de agua, enfríe, diluya a 500 ml y filtre.
Mida 25 ml de la infusión dentro de una cápsula de porcelana de 2 litros, adicione 20ml de la solución de índigo y 750ml de agua. Adicione la solución estándar de permanganato, 1 ml a la vez, hasta que cambie el color azul a verde, luego agregue otras gotas hasta que se torne de color amarillo dorado.
De manera similar, titule una mezcla de 20ml de solución de índigo y 750ml de agua. Multiplique la diferencia entre las titulaciones por el factor deseado para obtener ácido quercitánico u oxígeno absorbido.
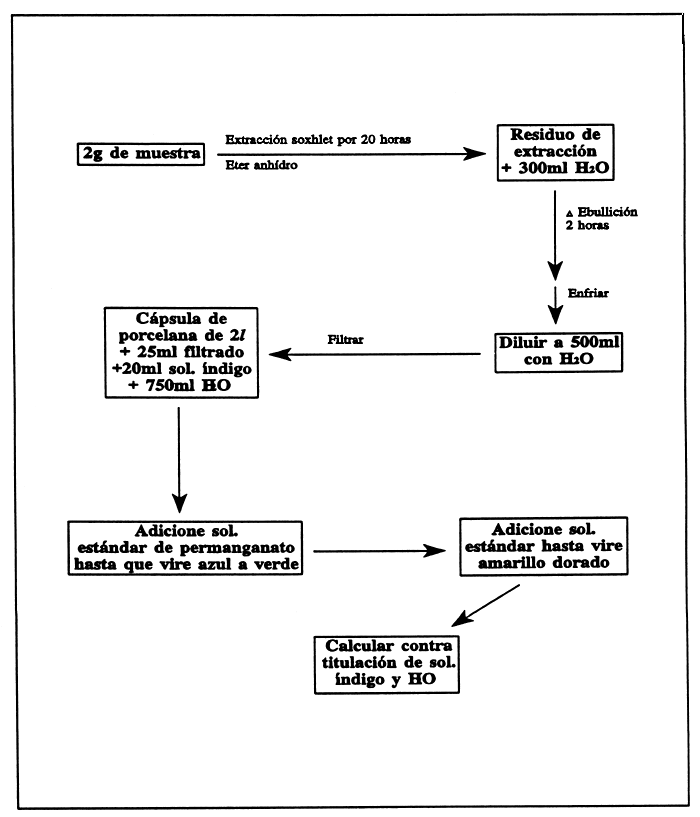
FIGURA 31
Método simple para la determinación de taninos en ingredientes alimenticios
El ácido fítico se puede encontrar en algunas semillas como ajonjolí, trigo, arroz, avena, sorgo, cebada, etc. Su presencia puede provocar deficiencias de fósforo en monogástricos por ligar y formar fitatos indigeribles que hacen indisponible ese elemento; afecta también la disponibilidad de calcio, magnesio o el fierro con los que forma el complejo fitina.
Reactivos
Materiales y equipo
Procedimiento
Coloque en un matraz erlemayer de 125 ml una muestra finamente molida (40 mallas), estimada para contener de 5 a 50 mg de fitato-P. Extraiga durante 30 min con 50 ml de solución al 3% de ATA bajo agitación constante.
Centrifuge la suspensión y transfiera una alícuota de 10 ml del sobrenadante a un tubo de centrífuga cónico. Agréguele 4 ml de la solución de FeCl3 preparada para contener 2 mg de Fe2 soplando rápidamente la pipeta.
Caliente durante 45 min el tubo con la muestra en un baño maría a 90–100°C. Si el sobrenadante no está claro después de 30 min adicione 1 ó 2 gotas de la sol. al 3% de sulfato de sodio en ATA y continúe con el calentamiento. Centrifuge por 10–15 min y decante con cuidado el sobrenadante. Lave dos veces el precipitado con 20–25 ml de solución de ATA al 3% dispersándolo bien y calentándolo en agua a ebullición por 5–10 min y centrifuge. Finalmente repita una vez el lavado con agua.
Disperse el precipitado en un poco de agua destilada, adicione 3 ml de la solución 1.5N de NaOH y mezcle. Lleve el volumen a aproximadamente 30 ml con agua destilada y caliente en agua a ebullición durante 30 minutos. Filtre en caliente (cuantitativamente) con un papel de retención moderada (Whatman 2 ó SyS 597).
Lave el precipitado con 60–70 ml de agua destilada caliente y deseche el filtrado. Si se desea hacer determinación de fósforo se puede usar el filtrado. Disuelva el precipitado en el papel con 40 ml de la solución 3.2N de HNO3 pasándolo a un matraz volumétrico de 100 ml. Lave varias veces el papel con agua destilada colectándola en el matraz. Enfríe la muestra a temperatura ambiente y afore con agua destilada.
Transfiera una alícuota de 5 ml a un matraz volumétrico de 100 ml y diluya a aproximadamente 70 ml. Agregue 20 ml de la solución 1.5M de KSCN y afore con agua destilada. Lea de inmediato (dentro de un lapso de 1 min) en el espectrofotómetro a 480 nm.
Corra un blanco de reactivos con cada muestra. Calcule el contenido de fierro a partir de un estándar de Fe(NO3)3 corrido al mismo tiempo o mediante una curva estándar preparada con anterioridad.
Cálculos
Calcule el fitato-P a partir de los resultados de fierro, asumiendo una tasa molecular de 4:6 fierro:fósforo
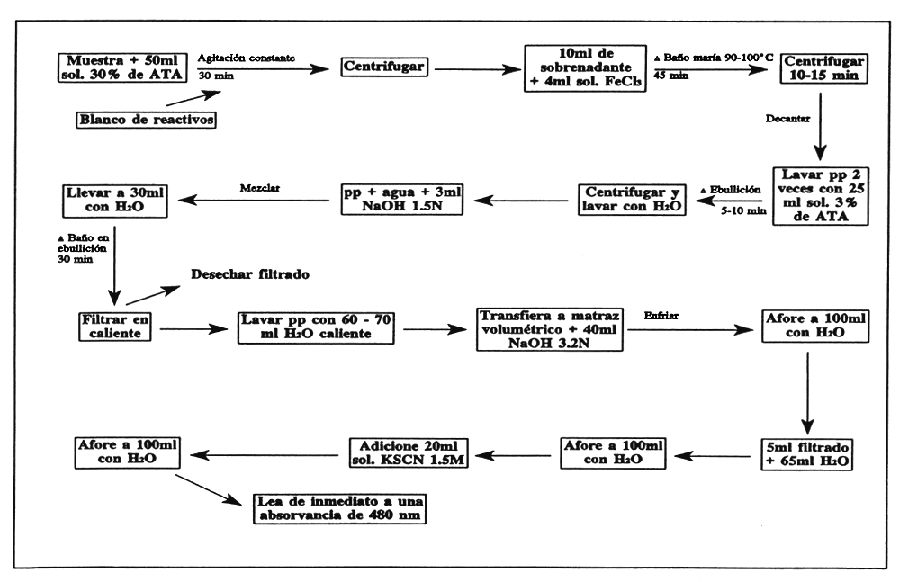
FIGURA 32
Método simple para la determinación de ácido fítico en ingredientes alimenticios.
Las saponinas se pueden encontrar en una gran variedad de plantas incluyendo la alfalfa, soya y semillas de leguminosas, habiéndose demostrado un efecto negativo sobre el crecimiento de monogástricos. Es un antinutriente estable al calor.
Reactivos
Materiales y equipo
Procedimiento
Muela finamente la muestra y séquela a peso constante. Pese alrededor de 40 g y colóquela a reflujo con acetona en el extractor soxhlet por al menos 24 horas para remover lípidos, pigmentos, etc. Cambie el solvente por metanol y continúe la extracción por al menos otras 24 horas. Deje enfriar el extracto metanólico y llévelo a 250 ml con metanol.
En este punto hay una modificación al método propuesta por Miriam Monforte (CICY, Mérida, Yucatán, México). En lugar de llevarlo a 250 ml como sugiere el método original, es más recomendable concentrar la muestra, lo que permite cuantificar cantidades muy pequeñas de saponina, para lo cual, pase el extracto metanólico a un rotoevaporador y concentre a sequedad. Resuspenda el residuo con un mínimo de metanol y páselo a un vial previamente pesado; si no se va a usar de inmediato, seque con nitrógeno y guárdelo en desecador. Pese el vial con la muestra seca y calcule el peso del residuo. Para utilizar la muestra, resuspenda con una cantidad conocida de metanol (alrededor de 1 ml).
Cromatografía cualitativa en capa fina
Coloque puntos del extracto sobre una placa de gel de sílica y revélelos con una solución n-butanol-etanol-amoníaco concentrado (3.5:1:2.5). Se pueden aplicar tres diferentes métodos para identificar las manchas de saponina: (1) aspersíon con ácido acético-anisaldeido; (2) tratamiento con nitrato de plata seguido de hidróxido de sodio y (3) tratamiento con una suspensión de eritrocitos en buffer isotónico. Para mayores detalles consulte a Fenwick y Oakenfull (1981).
Cromatografía cuantitativa en capa fina
Coloque series de puntos con volúmenes conocidos del extracto y de una solución estándar de saponina sobre las placas de cromatografía. Los puntos deben ser colocados de tal manera que cada punto de extracto vaya al lado de los de estándar de saponina. Revele las placas de la misma manera que en el apartado anterior y las manchas deben ser visualizadas mediante la aspersíon de una solución de ácido sulfúrico en metanol, seguida de calentamiento a 110°C por 30 min. Mida la intensidad de las manchas de saponina por medio de un densitómetro y calcule las áreas de los picos en el graficador con un planímetro. los resultados son expresados como la relación (R) de las áreas de los picos de la muestra desconocida con respecto a la del estándar.
Se ha determinado que la concentración de saponina en la muestra (en μg) es proporcional al cuadrado de las áreas relativas (R2). Grafique R2 contra el volumen de la gota de extracto metanólico colocada en la placa. La pendiente de la línea (calculada por el método de cuadrados mínimos), divido entre la pendiente de una línea obtenida de una curva patrón, dará la concentración de la saponina en el extracto y de esa manera, el contenido de saponina en la muestra. El método puede tener un error experimental acumulado de ±20%.
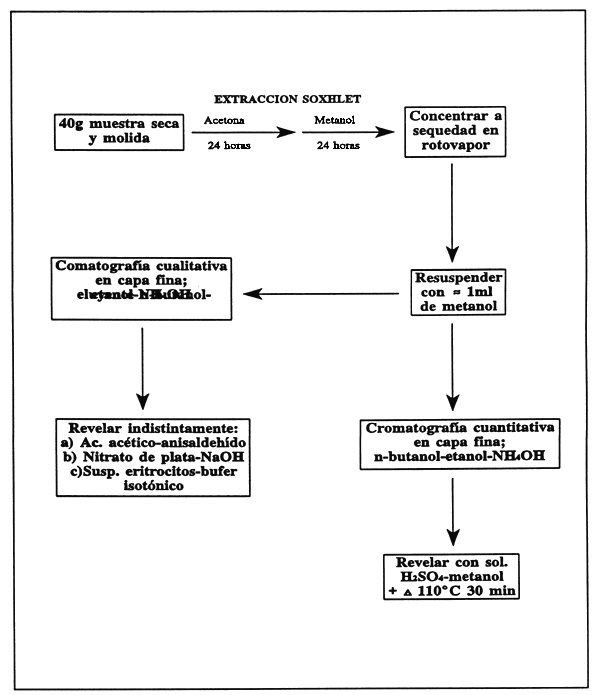
FIGURA 33
Determinación de saponinas en ingredientes alimenticios.
![]()
![]()
![]()