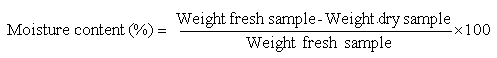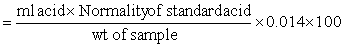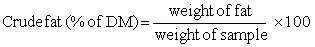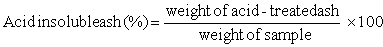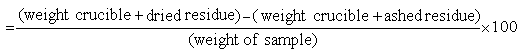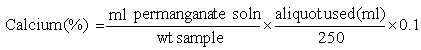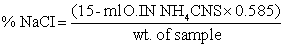![]()
![]()
![]()
INTRODUCTION
The following methods of feed analysis are provided by courtesy of the Tropical Products Research Institute, London, England and were prepared by Cockerell, Halliday and Morgan (1975). In addition to the techniques reproduced here the original text contains methods for the determination of thioglucoside, urease, total sugars in molasses, potassium content of molasses, and free gossypol in cottonseed meal.
A footnote has been added to the method for crude fat analysis, based on the experience of the author of this manual. The method for calculating NFE has been altered, so that it conforms with other parts of this manual.
The methods of analysis are otherwise as published by the original authors, who are thanked for their permission to reproduce them here.
SAMPLE PREPARATION
All samples of raw materials and finished products should be stored for analysis in sealed containers. Prior to analysis, samples should be ground to pass through a 1 mm screen; where grinding is difficult a mortar and pestle can be used. In the case of moist samples or where grinding is unduly prolonged moisture determinations should be carried oat on the material before and after grinding, as described below.
MOISTURE DETERMINATION
Weigh out 4 to 5 g of the sample in a covered, flat, aluminium dish and dry to constant weight at 100° C. preferably in an oven fitted with controlled ventilation.
CRUDE PROTEIN
Total nitrogen is determined by the Kjeldahl method and the result multiplied by 6.25 to give crude protein.
Reagents:
1. Sulphuric acid (98%), nitrogen free.
2. Potassium sulphate, reagent grade.
3. Mercuric oxide, reagent grade.
4. Paraffin wax.
5. Sodium hydroxide, 40% solution.
6. Sodium sulphide, 4% solution.
7. Pumice chips.
8. Boric acid/indicator solution. Add 5 ml of indicator solution (0.1% methyl red and 0.2% bromocresol green in alcohol) to 1 litre saturated boric acid solution.
9. Hydrochloric acid standard solution (0.1N).
Apparatus:
1. Macro Kjeldahl digestion and distillation units.
2. Kjeldahl flasks (500 ml capacity or larger).
3. Conical flasks, 250 ml.
Method:
Weigh accurately approximately 1 g of sample into a digestion flask and add 10 g potassium sulphate, 0.7 g mercuric oxide (pre-measured catalyst tablets containing these two reagents are available), and 20 ml sulphuric acid. Heat the flask gently at an inclined angle until frothing subsides and then boil until the solution clears.Continue boiling for a further half hour. If the frothing is excessive, a small amount of paraffin wax can be added.
On cooling add about 90 ml distilled water, recool, add 25 ml sulphide solution and mix. Add a small piece of pumic to prevent 'bumping' and 80 ml of sodium hydroxide solution while tilting the flask so that two layers are formed. Connect rapidly to the condenser unit, heat and collect distilled ammonia in 50 ml boric acid/indicator solution. Collect 50 ml of distillate. On completion of distillation remove the receiver (wash condenser tip) and titrate against standard acid solution.
Calculation:
Nitrogen content of sample (%)
Crude protein content (%) = nitrogen content × 6.25
CRUDE FAT
Reagents and equipment:
1. Petroleum ether (b.p. 40-60°C).
2. Extraction thimbles.
3. Soxhlet extraction apparatus.
Method:
Weigh into an extraction thimble 2-3 g of the dried sample (residue from dry matter determination can be used). Place the thimble inside the soxhlet apparatus. Place a dry, tared solvent flask in position beneath, add the required quantity of solvent and connect to condenser. Adjust heating rate to give a condensation rate of 2 to 3 drops/second and extract for 16 hours. (The extraction time may be reduced to a minimum of six hours by increasing the condensation rate). 1/ On completion remove the thimble and reclaim ether using the apparatus. Complete the removal of ether on a boiling water bath and dry flask at 105° C for 30 minutes. Cool in a desiccator and weigh.1/Author's Note: If the straight type of extraction tube is used to hold the thimble instead of a soxhlet apparatus, extraction times can be reduced to one hour and still provide results accurate enough for routine work. In this way nine samples can be analysed in duplicate per day, using only 3 heaters. This is important as lipid analysis is often a limiting factor in obtaining the rapid results necessary for timely quality control; lack of equipment prevents analysis of more than a few samples if 16, or even 6, hours extraction time is insisted on.
Calculation:
ASH
Weigh a 2 g sample into a dry, tared porcelain dish and then place in a muffle furnace at 600° C for 6 hours. Cool in a desiccator and weigh.
Calculation:
ACID SOLUBLE AND INSOLUBLE ASH
Reagents and Apparatus:
1. Hydrochloric acid (1 to 2.5 v/v)
2. Filter paper, ashless
3. Dishes, porcelain
Method:
Use the residue obtained from ash determination. Boil with 25 ml hydrochloric acid, taking care to avoid spattering, filter through ashless paper and wash with hot water until acid free. Place filter paper and residue into a dry, tared porcelain dish and place in a muffle furnace at 600 C for 2 hours or until carbon free.
Calculation:
CRUDE FIBRE
Crude fibre is determined as that fraction remaining after digestion with standard solutions of sulphuric acid and sodium hydroxide under carefully controlled conditions.
Reagents:
1. Sulphuric acid solution (0.255N).
2. Sodium hydroxide solution (0.313N).
3. Antifoam reagent (Octyl alcohol).
4. Ethyl alcohol.
5. Hydrochloric acid, 1% v/v.
Apparatus:
1. Beakers, 600 ml tall-sided.
2. Round-bottom flask condenser unit.
3. Buchner flasks, 1 litre.
4. Buchner funnels, Hartley 3 section pattern.
5. Crucibles, silica with porous base.
6. Rubber cones to fit above.
Method:
Weigh about 2 g of the dried, fat-free sample into a 600 ml beaker (for convenience the residue from ether extraction can be used). Add 200 ml of hot sulphuric acid, place the beaker under the condenser and bring to boiling within 1 minute. Boil gently for exactly 30 minutes, using distilled water to maintain volume and to wash down particles adhering to the sides. Use antifoam if necessary. Filter through Whatman No. 541 paper in a Buchner funnel using suction and wash well with boiling water. Transfer residue back to beaker and add 200 ml hot sodium hydroxide solution. Replace under the condenser and again bring to boil within 1 minute. After boiling for exactly 30 minutes filter through porous crucible and wash with boiling water, 1% hydrochloric acid and then again with boiling water. Wash twice with alcohol, dry overnight at 100 °C, cool and weigh. Ash at 500 °C for 3 hours, cool and weigh. Calculate the weight of fibre by difference.
Calculation:
Crude fibre (% of fat-free DM)
NITROGEN-FREE EXTRACT (NFE)
Calculate as 100 - % crude protein - 7, crude fat - % crude fibre - % ash - % moisture.
FREE FATTY ACID CONTENT OF FATS & OILS
Reagents and apparatus:
1. Ethyl alcohol.
2. Phenolphthalein (1% soln. in alcohol).
3. Sodium hydroxide (0.25N).
4. Stoppered flasks, 250 ml.
Method:
Weigh 7.05 g oil or fat into a stoppered flask, add 50 ml alcohol previously neutralized by adding sufficient 0.25N sodium hydroxide to give faint pinkish colour with phenolphthalein (2 ml). Titrate with sodium hydroxide with vigorous shaking until a permanent faint pink colour appears and persists.
Calculation:
Free fatty acids % (as oleic acid) = volume of 0.25N NaOH used in titration
CALCIUM
Reagents:
1. Hydrochloric acid (1 to 3 v/v).
2. Nitric acid (70%).
3. Ammonium hydroxide (1 to 1 v/v).
4. Methyl red indicator (Dissolve 1 g in 200 ml alcohol),
5. Ammonium oxalate (4.2% soln.).
6. Sulphuric acid (98%).
7. Standard potassium permanganate soln. (0.05N).
Apparatus:
1. Porcelain dishes.
2. Volumetric flasks, 250 ml.
3. Beakers, 250 ml.
4. Quantitative filter paper & funnels.
5. Burette.
Method:
Weigh 2.5 g finely ground material into a porcelain dish and ash as above (alternatively use residue from ash determination). Add 40 ml hydrochloric acid and a few drops of nitric acid to the residue, boll, cool and transfer to a 250 ml volumetric flask. Dilute to volume and mix.Pipette a suitable aliquot of the solution (100 ml for cereal feeds; 25 ml for mineral feeds) into a beaker, dilute to 100 ml and add 2 drops of methyl red. Add ammonium hydroxide dropwise until a brownish orange colour is obtained, then add two drops of hydrochloric acid to give a pink colour. Dilute with 50 ml water, boil and add while stirring 10 ml of hot 4.2% ammonium oxalate solution. Adjust pH with acid to bring back pink colour if necessary. Allow precipitate to settle out, and filter, washing precipitate with ammonium hydroxide solution (1 to 50 v/v). Place the filter paper with precipitate back in beaker and add a mixture of 125 ml water and 5 ml sulphuric acid. Heat to 70° C and titrate against the standard permanganate solution.
Calculation:
SODIUM CHLORIDE
Reagents
1. Standard 0.1N silver nitrate solution.
2. Standard 0.1N ammonium thiocyanate solution.
3. Ferric indicator - saturated aqueous solution of ferric alum.
4. Potassium permanganate solution - 6% w/v.
5. Urea solution - 5% w/v.
6. Acetone (A.R. grade).
Method:
Weigh 2 g sample into a 250 ml conical flask. Moisten sample with 20 ml water and then add, by pipette, 15 ml 0.1N silver nitrate solution - mix well. Add 20 ml concentrated nitric acid and 10 ml potassium permanganate solution and mix. Heat mixture continuously until liquid clears and nitrous fumes are evolved - cool. Add 10 ml urea solution and allow to stand for 10 minutes. Add 10 ml acetone and 5 ml ferric indicator and back titrate the excess silver nitrate with the 0.1N thiocyanate solution to the red brown end point.
Calculation:
Calculate result as sodium chloride.
PHOSPHOROUS
Reagents:
1. Molybdovanadate reagent. Dissolve 40 g ammonium molybdate 4H 0 in 400 ml hot water and cool. Dissolve 2 g ammonium metavanadate in 250 ml hot water, cool and add 450 ml 70% perchloric acid. Gradually add the molybdate to the vanadate solution with stirring and dilute to 2 litres.2. Phosphorous standards. Prepare stock solution by dissolving 8.788 g potassium dihydrogen orthophosphate in water and making up to 1 litre. Prepare the working solution by diluting the stock 1 in 20 (working concentration = 0.1 mg P/ml).
Apparatus:
1. Spectrophotometer to read at 400 mm.
2. Graduated flasks, 100 ml.
Method:
Pipette an aliquot of the sample solution prepared as for the calcium determination into a 100 ml flask and add 20 ml of the molybdovanadate reagent. Make up to volume, mix and stand for 10 minutes. Transfer aliquots of the working standard containing 0.5, 0.8, 1.0 and 1.5 mg phosphorus to 100 ml flasks and treat as above. Read sample at 400 mm setting the 0.5 mg standard at 100% transmission. Determine mg phosphorus in each sample aliquot from a standard curve.
CAROTENE
Reagents:
1. Acetone.
2. Hexane (b.p. 60-70°C).
3. Diatomaceous earth.
4. Activated magnesia.
Apparatus:
1. Reflux apparatus.
2. Chromatographic tubes (30 cm × 12 mm i.d.).
3. Spectrophotometer.
Weigh out 2 g of sample (more or less depending on carotene content) and reflux with 15 ml of a 3:7 acetone:hexane mixture for one hour. Cool and filter into a 50 ml volumetric flask, washing with hexane and making up to volume.
Prepare a column using a 1:1 mixture of activated magnesia and diatomaceous earth. Pack under vacuum to a total depth of about 7 cm and then add 0.5 cm anhydrous sodium sulphate to the top of the column. All materials used in making the column must be thoroughly dry.
With the column under vacuum and a 50 ml volumetric flask to collect the eluate apply 25 ml of the sample extract to the column. As the last of the extract enters the adsorbent add acetone:hexane eluent (1:9) and continue until the carotene band is washed through. Add acetone:hexane eluent (1:9) to the flask to make up to volume and then read in the spectrophotometer at 436mm . Prepare a standard curve using beta carotene concentrations of 0.1 to 5m g/ml of acetone hexane mixture.
AFLATOXIN ANALYSIS
A method of aflatoxin analysis is outlined below which is suitable for materials such as groundnut meal, coconut meal and palm kernel meal. For full details of the method and for alternative procedures reference should be made to B.D. Jones (1972), Methods of aflatoxin analysis: Report No G 70, Tropical Products Institute, London, England.
Reagents:
1. Chloroform (Reagent grade).
2. Diethyl ether (Reagent grade).
3. Chloroform/methanol mixture (95/5 v/v).
4. 'Celite' diatomaceous earth.
5. Kieselgel 'G' (Merck).
6. Qualitative standard. Helps to distinguish aflatoxin spots from other fluorescent spots which may be present. A groundnut meal containing aflatoxins B, obtainable from the Tropical Products Institute, London, can be used for this purpose.
Apparatus:
1. Thin layer chromatography plates, 20 × 20 cm.
2. U.V. lamp, peak emission at 365 mu.
3. Bottles, wide-mouthed, 250 ml.
4. Micropipettes.
5. Shaking device.
Method:
Weigh 10 ml of material into a wide mouthed bottle and thoroughly mix in 10 ml of water. (If high fat material is used, a prior Soxhlet extraction with petroleum ether will be necessary). Add 100 ml of chloroform, stopper with a chloroform resistant bung and shake for 30 minutes. Filter the extract through 'Celite', take 20 ml of filtrate and make up to 25 ml (Soln. A.). Take another 20 ml of filtrate and concentrate to 5 ml (Soln. B.).Prepare thin layer plates by shaking Kieselgel 'G' (100 g) with water (220 ml) for 20 minutes and applying the mixture to the plates with a suitable apparatus to a depth of 508 u. Leave for 1 hour, then dry at 100°C. Spot 10 and 20 ul of solution B and 5 and 10 ul of solution A onto a plate, together with a qualitative standard spot, in a line 2 cm from the bottom of the plate and at least 2 cm in from each side. Carry out the spot application in subdued light.
Develop the plate in diethyl ether to a height of 12 cm. Allow to dry in subdued light then redevelop the plate in chloroform:methanol (95/5, v/v) to a height of 10 cm from the baseline. Examine the plate in a dark room, 30 cm from the UV source. The presence of a blue fluorescent spot at Rf 0.5 to 0.55 indicates aflatoxin B (check that the standard spot also lies in this range). The presence of a second spot at Rf. 0.45 to 5 indicates aflatoxin G. The toxicity level of a sample can then be classified in terms of aflatoxins B and G according to the following table:
|
Vol. Applied (ul) |
Cone. of aflatoxins (m g/kg) |
Toxicity level if fluorescence observed | |
|
|
No fluorescence |
Fluorescence |
|
|
5m l (Soln. A) |
< 1 000 |
> 1 000 |
very high |
|
10m l (Soln. A) |
< 500 |
500 - 1 000 |
high |
|
10m l (Soln. B) |
< 100 |
100 - 500 |
medium |
|
20m l (Soln. B) |
< 50 |
50 - 100 |
low |
Note: References for each method quoted in this appendix are given in the original paper (Cockerell, et al., 1975).
![]()
![]()
![]()