![]()
![]()
![]()
AVANT-PROPOS
L'objectif des présentes directives est d'aider à assurer la fiabilité des résultats d'analyse en vérifiant la conformité avec les limites maximales de résidus se trouvant dans les aliments faisant l'objet d'un commerce international. Des résultats d'analyse fiables sont essentiels pour protéger la santé des consommateurs et faciliter le commerce international.
Outre les présentes directives, il existe d'autres recommandations pertinentes du Codex élaborées par le Comité du Codex sur les résidus de pesticides dans le domaine de l'application des limites maximales de résidus, à savoir:
1 Méthodes d'échantillonnage recommandées pour la détermination des résidus de pesticides (réf.: CAC/VOL XIII - deuxième édition, Partie VI ou CAC/PR 5-1984), telles qu'amendées pour ce qui concerne la viande et la volaille (ALINORM 91/40; voir aussi ALINORM 89/24A, Annexe II et ALINORM 91/24A Annexe VIII).DIRECTIVES CONCERNANT LES BONNES PRATIQUES DE LABORATOIRE EN MATIÈRE D'ANALYSE DES RÉSIDUS DE PESTICIDES2 Portion des produits à laquelle s'appliquent les limites maximales de résidus et qui est soumise à l'analyse (réf.: CAC/VOL XIII - Ed. 1, Partie V ou CAC/PR6-1984).
3 Notes explicatives sur les limites maximales de résidus pour des résidus de pesticides (réf.: CAC/VOL XIII - première édition, Partie III).
4 Recommandations concernant les méthodes d'analyse pour les résidus de pesticides (réf.: CAC/VOL XIII - deuxième édition, Partie VIII ou CAC/PR 8-1984)
5 Classification Codex des aliments destinés à l'alimentation humaine et animale (réf.: CAC/PR4-1989)
1. INTRODUCTION
Le document du Codex ALINORM 76/24 Annexe IV (Rapport du groupe de travail ad hoc sur les méthodes d'analyse) contenait l'énoncé ci-après:
"L'objectif ultime des pratiques loyales en matière de commerce international dépend, entre autres, de la fiabilité des résultats de l'analyse. Ce facteur est conditionné à son tour - notamment lorsqu'il s'agit du dosage des résidus de pesticides -non seulement par l'existence de méthodes d'analyse sûres, mais aussi par la compétence de l'analyste et le respect des bonnes pratiques en matière d'analyse des résidus de pesticides".Les présentes directives définissent ces bonnes pratiques d'analyse et sont examinées en trois volets interdépendants:
L'analyste (section 2);
Ressources de base (section 3);
L'analyse (section 4).
Les spécifications concernant les installations, la gestion, le personnel, l'assurance et le contrôle de la qualité, la documentation des résultats et les données brutes, ainsi que les thèmes connexes, qui sont considérés comme des conditions préalables pour obtenir des résultats fiables et vérifiables sont décrites en général dans la norme ISO/IEC 17025 (1999) et dans une série de documents directifs sur les bonnes pratiques de laboratoire de l'OCDE, ainsi que dans les lois et règlements nationaux correspondants. Ces directives Codex, qui ne sont pas exhaustives, décrivent les principes et les pratiques essentiels à suivre dans l'analyse des résidus de pesticides.
2. L'ANALYSTE
2.1 L'analyse des résidus consiste en une série d'opérations dont les modalités sont le plus souvent bien connues, ou faciles à comprendre par un chimiste qualifié, mais du fait que les concentrations sont de l'ordre de µg à mg/kg et que les analyses sont parfois difficiles, il est indispensable de veiller à tous les détails. L'analyste responsable doit avoir les qualifications professionnelles, l'expérience et les compétences requises en matière d'analyse des résidus. Le personnel doit être entraîné au maniement correct des appareils et aux techniques de laboratoire de base. En outre, l'analyste qui utilise la méthode pour la première fois doit effectuer les essais décrits aux sections 4.4.5 du tableau 4 pour montrer qu'il peut utiliser la méthode en suivant les paramètres de performance prévus et établis durant la validation de la méthode avant d'appliquer la méthode pour l'analyse des échantillons. Il doit être bien informé des principes de l'analyse des résidus et des exigences des systèmes d'assurance de la qualité de l'analyse. Il doit par ailleurs connaître l'objet de chaque étape de la méthode utilisée et savoir qu'il est important de se conformer exactement aux méthodes prescrites et de noter tout écart inévitable. Il doit également être formé à l'évaluation et à l'interprétation des données qu'il produit. Il sera bon de noter sur un registre la formation et l'expérience de tous les membres du personnel.
2.2 Lorsque l'on installe un laboratoire d'analyse des résidus, une partie de la période de formation du personnel devrait se passer dans un laboratoire bien rodé où aide consultative et formation seraient dispensées par des personnes expérimentées. Si le laboratoire est appelé à analyser une large gamme de résidus de pesticides, le personnel devra peut-être se perfectionner dans plusieurs laboratoires déjà en service.
3. RESSOURCES DE BASE
3.1 Le laboratoire
3.1.1. Le laboratoire et ses installations devraient être conçus de manière que les opérations puissent être réparties entre des zones bien définies, en vue de garantir un maximum de sécurité et un minimum de risques de contamination des échantillons. Les installations de chaque zone devraient être construites en matériaux résistants aux produits chimiques dont l'utilisation paraît probable. Ainsi, il faudrait théoriquement installer des pièces distinctes pour la réception et le stockage des échantillons, pour la préparation des échantillons, pour l'extraction et la séparation et pour le rangement des instruments qui serviront à l'analyse elle-même. La zone destinée aux opérations d'extraction et de purification devrait être conforme aux normes établies pour les laboratoires utilisant des solvants et toutes les installations d'évacuation des vapeurs et fumées devraient être d'excellente qualité. La réception, le stockage et la préparation des échantillons devraient avoir lieu dans des zones réservées exclusivement à l'analyse des résidus. Il faudra en priorité garantir l'intégrité des échantillons et la sécurité personnelle.
3.1.2 La sécurité du laboratoire doit aussi être envisagée sous l'angle de ce qui est nécessaire et souhaitable car il faut reconnaître que les normes très strictes, appliquées dans les laboratoires d'analyse des résidus de certains pays du monde en ce qui concerne les conditions de travail, seraient totalement irréalistes ailleurs. Il doit être interdit de fumer, de manger, de boire et de se maquiller dans la zone de travail. De petites quantités seulement de solvants doivent y être conservées et la majeure partie de ces produits doit être stockée dans une pièce séparée, éloignée de la zone de travail principale. Il convient d'éviter chaque fois que possible l'emploi de solvants et de réactifs ayant des propriétés toxiques aiguës ou chroniques. Tous les solvants usés doivent être gardés en lieu sûr et évacués en toute sécurité et sans porter atteinte à l'environnement, en tenant compte des éventuels règlements nationaux en vigueur.
3.1.3 La zone de travail principale doit être conçue et équipée pour l'utilisation d'une gamme appropriée de solvants destinés à l'analyse. Tout le matériel tel que dispositifs d'éclairage, macérateurs et réfrigérateurs doit être d'un type ne produisant ni étincelles ni explosions. Les opérations d'extraction, de purification et de concentration doivent être menées dans un espace bien ventilé, de préférence dans des hottes fermées.
3.1.4 Il convient d'utiliser des écrans protecteurs lorsque la verrerie est employée sous vide ou sous pression. Il y a lieu de prévoir un ample stock de lunettes protectrices, de gants et autres vêtements de protection, des installations pour se laver en cas d'urgence et les fournitures nécessaires pour le traitement des débordements. Un matériel adéquat de lutte contre les incendies est également indispensable. Le personnel doit savoir que de nombreux pesticides ont des propriétés toxiques aiguës ou chroniques et qu'il faut donc manipuler les produits étalons avec beaucoup de précautions.
3.2 Matériel et fournitures
3.2.1 Le laboratoire doit être bien alimenté en électricité et en eau. Des réserves suffisantes de réactifs, de solvants, de verrerie pour les gaz, de chromatographes, etc. sont indispensables.
3.2.2 Il faut assurer l'entretien des chromographes, balances, spectrophotomètres, etc., valider régulièrement leurs performances et consigner dans un registre toutes les interventions d'entretien ou de réparation. L'étalonnage est indispensable pour la mesure des performances du matériel. Des courbes d'étalonnage et des comparaisons avec les substances étalons peuvent suffire.
3.2.3 II faudra procéder à l'étalonnage et au ré-étalonnage réguliers du matériel uniquement lorsque le changement éventuel dans la valeur nominale peut contribuer sensiblement à l'incertitude de la mesure. Les balances et les pipettes/distributeurs automatiques et matériel similaire doivent être régulièrement étalonnés. On vérifiera à intervalles réguliers les températures de fonctionnement des réfrigérateurs et des congélateurs. Toutes les données doivent être consignées.
3.2.4 En théorie, l'équipement devrait être modernisé régulièrement pour suivre l'évolution des techniques, mais en pratique il suffit qu'il soit assez perfectionné pour le travail demandé.
3.2.5 Tous les laboratoires doivent disposer d'un assortiment suffisant de pesticides de référence ayant un degré de pureté connu et acceptable. Celui-ci doit comprendre tous les composés initiaux dont le laboratoire surveille les concentrations dans des échantillons de produits, ainsi que leurs métabolites pour lesquels des LMR ont été fixées.
3.2.6 Les produits de référence, les solutions de réserve et les réactifs doivent tous porter une étiquette indiquant clairement la date limite d'utilisation et être stockés dans de bonnes conditions. Les composés de référence "purs" doivent être conservés de manière à minimiser le taux de dégradation, par exemple à basse température, dans un endroit sec et obscur. On veillera également à ce que les solutions de pesticides de référence ne soient pas décomposées sous l'effet de la lumière ou de la chaleur durant le stockage ou ne deviennent concentrées du fait de l'évaporation des solvants.
4. L'ANALYSE
Les méthodes appliquées pour le dosage des résidus de pesticides devraient en général satisfaire aux critères présentés au tableau 3.
4.1 Risques de contamination
4.1.1 L'analyse des résidus de pesticides diffère considérablement de la micro-analyse en raison du problème de la contamination et des interférences. Des quantités infimes de contaminants dans les échantillons finals utilisés pour le dosage peuvent donner lieu à des erreurs telles que des faux résultats positifs et des faux résultats négatifs, et causer une perte de sensibilité, ce qui peut empêcher l'analyste de détecter le résidu. La contamination peut être imputable à tout ce qui sert à l'échantillonnage, au transport et au stockage des échantillons, comme aux analyses. Il faut vérifier avant usage que la verrerie, les réactifs, les solvants organiques et l'eau ne contiennent pas de contaminants, en procédant à l'analyse d'un blanc de réactifs.
4.1.2 Les produits d'entretien, les crèmes protectrices, les savons contenant des antiseptiques, les insecticides en aérosols, les parfums et les cosmétiques peuvent tous causer des problèmes d'interférence et sont particulièrement gênants lorsqu'on utilise un détecteur à capture électronique. Il n'y a pas d'autre solution au problème que d'interdire l'utilisation de ces produits dans le laboratoire.
4.1.3 Les lubrifiants, les produits de scellement, les matières plastiques, les caoutchoucs naturels et synthétiques, les gants de protection, l'huile provenant des conduites d'air et les impuretés de fabrication dans les cuvettes d'extraction, les papiers filtres et l'ouate peuvent aussi contaminer la solution finale à tester.
4.1.4 Les réactifs chimiques, les adsorbants et les solvants utilisés en laboratoire peuvent renfermer, adsorber ou absorber des constituants susceptibles de gêner l'analyse. Il faut parfois purifier les réactifs et les adsorbants et il est généralement nécessaire d'utiliser des solvants redistillés. L'eau déionisée est souvent suspecte et on lui préférera l'eau redistillée. L'eau du robinet ou celle d'un puits sont souvent utilisables.
4.1.5 La contamination de la verrerie, des seringues et des colonnes de chromatographie gazeuse peut provenir d'échantillons ou d'extraits précédents. Toute la verrerie doit être nettoyée avec un détergent, rincée à fond avec de l'eau distillée (ou autre eau propre), puis rincée avec le solvant qui sera utilisé. La verrerie qui servira à l'analyse des traces doit être gardée séparément et ne pas être utilisée à d'autres fins.
4.1.6 II faudrait toujours conserver les pesticides de référence à une température appropriée dans une pièce séparée du laboratoire principal. Les solutions et extraits concentrés de référence devraient être placés ailleurs.
4.1.7 II faut se méfier des appareils contenant du polychlorure de vinyle (PVC) et, s'il a été démontré qu'ils sont une source de contamination, il faut en interdire l'introduction dans le laboratoire d'analyse des résidus. D'autres matières contenant des plastifiants sont également suspectes, mais le PTFE et les caoutchoucs de silicone sont en général acceptables, ainsi que d'autres produits dans certaines conditions. Les récipients de stockage des échantillons peuvent provoquer une contamination, aussi faut-il toujours utiliser des flacons en verre munis de bouchons rodés. Les instruments d'analyse devraient toujours être rangés dans une pièce séparée. La nature et l'importance de la contamination peuvent varier selon les techniques de détermination utilisées et les concentrations de résidus de pesticides à déterminer. On peut réduire ces problèmes de contamination, qui sont importants si l'on utilise la chromatographie gazeuse ou la chromatographie liquide à haute résolution en complétant la détermination par une analyse spectrophotométrique ou inversement. Si les concentrations de résidus sont relativement élevées, les interférences dues aux solvants et à d'autres substances peuvent être insignifiantes par rapport à la quantité de résidus présente, mais de nombreux problèmes peuvent être résolus à l'aide de détecteurs spécifiques. En outre, si le contaminant ne gêne pas la détermination du résidu recherché, sa présence peut être tolérée.
4.1.8 Les analyses de résidus et de préparations doivent se faire dans des installations de laboratoire complètement séparées. Les échantillons et la préparation des échantillons doivent être séparés de toutes les opérations de laboratoire de résidus afin d'empêcher toute contamination croisée.
4.2 Réception et stockage des échantillons
4.2.1 Chaque échantillon reçu en laboratoire devrait être accompagné de renseignements sur l'échantillon, sur l'analyse requise et sur les dangers potentiels associés à sa manipulation.
4.2.2 A la réception de l'échantillon, il faut lui attribuer immédiatement un code d'identification unique qui l'accompagnera à toutes les étapes de l'analyse jusqu'à la présentation des résultats. Si possible, on utilisera un système approprié de contrôle de l'élimination des échantillons, pour lequel on tiendra un registre.
4.2.3 La transformation des échantillons et le sous-échantillonnage devraient être effectués à l'aide de procédés qui fournissent des portions d'essai représentatives et n'ont pas d'effet sur la concentration de résidus présents.
4.2.4 Si des échantillons ne peuvent être analysés immédiatement mais doivent être analysés rapidement, ils doivent être réfrigérés à une température de 1 à 5°C, à l'abri de la lumière solaire directe et analysés dans les jours qui suivent. Toutefois, les échantillons reçus surgelés doivent être conservés à £ -16 °C jusqu'à l'analyse. Dans certains cas, il peut être nécessaire de stocker les échantillons pendant une période plus longue avant l'analyse. Les échantillons doivent être stockés à environ -20°C, température à laquelle la dégradation enzymatique des résidus de pesticides est habituellement très lente. Si l'on ne peut éviter un stockage prolongé, on en vérifiera les effets en analysant des échantillons enrichis dans les mêmes conditions et pendant le même laps de temps. Des informations utiles sur la stabilité au stockage des résidus de pesticides figurent dans les publications annuelles de la FAO intitulées Résidus de pesticides - Evaluations préparées par la JMPR FAO/OMS, complétées par les renseignements fournis par les fabricants pour appuyer l'homologation de leurs pesticides.
4.2.5 Lorsqu'il faut congeler des échantillons, il est recommandé de prélever des portions d'essai avant la congélation afin de réduire au minimum l'effet possible de la séparation de l'eau en cristaux de glace durant le stockage. Il faudra aussi faire en sorte que toute la portion d'essai soit utilisée dans l'analyse.
4.2.6 Ni les récipients utilisés pour le stockage ni leurs couvercles ou bouchons ne doivent permettre une fuite de ou des analyte(s) du compartiment de stockage. Les récipients ne doivent pas fuir. Tous les échantillons doivent être étiquetés clairement et de manière indélébile et être enregistrés dans un répertoire. Les extraits et les solutions d'épreuve finales ne doivent pas être exposés directement au soleil.
4.3 Protocoles normalisés
4.3.1 II faut utiliser des protocoles normalisés pour toutes les opérations. Ceux-ci doivent contenir des instructions de travail complètes ainsi que des informations sur l'applicabilité, les performances prévues, les exigences en matière de contrôle de qualité interne (vérification des performances) et le calcul des résultats. Ils devraient également contenir des informations sur tout danger découlant de la méthode, des produits étalons ou des réactifs.
4.3.2 Tout écart par rapport à un protocole normalisé doit être autorisé par l'analyste responsable et consigné.
4.4 Validation des méthodes[24]
4.4.1 On entend par méthode d'analyse la série de procédés à suivre depuis la réception d'un échantillon jusqu'à la production du résultat final. La validation est le processus permettant de vérifier qu'une méthode est adaptée au but recherché. La méthode peut être élaborée sur place, prise dans la documentation ou obtenue auprès d'un tiers. Elle sera ensuite adaptée ou modifiée afin de répondre aux exigences et aux capacités du laboratoire et/ou au but dans lequel la méthode sera utilisée. En général, la validation a lieu une fois que l'élaboration de la méthode est terminée et l'on suppose que des spécifications telles que l'étalonnage, la pertinence du système, la stabilité de l'analyte, etc, ont été établis de manière satisfaisante. Lorsqu'on valide et qu'on utilise une méthode d'analyse, il faut prendre des mesures dans la fourchette étalonnée du système de détection utilisé. Généralement, la validation précède l'application pratique de la méthode à l'analyse des échantillons, mais la vérification de performance ultérieure est un aspect de continuité important du processus. Les spécifications concernant les données de vérification des performances constituent une sous-série de celles requises pour la validation de la méthode.
Les essais d'efficacité (ou d'autres méthodes d'essais inter-laboratoires), chaque fois que possible, constituent un important moyen de vérifier l'exactitude générale des résultats fournis par une méthode et donnent des informations sur la variabilité des résultats d'un laboratoire à l'autre. Toutefois, les essais d'efficacité ne portent pas habituellement sur la stabilité ou l'homogénéité de l'analyte ni sur l'extractabilité des analytes dans l'échantillon traité.
Lorsque des données sur l'incertitude sont nécessaires, elles doivent inclure des données sur la vérification des performances et ne pas s'appuyer uniquement sur les données de validation de la méthode.
4.4.2 Chaque fois qu'un laboratoire entreprend d'élaborer et/ou de modifier une méthode, les effets des variables analytiques devraient être établis, par exemple en recourant à des essais de robustesse avant la validation. Des contrôles rigoureux doivent être effectués en respectant tous les aspects de la méthode qui peuvent influer sur les résultats tels que: taille des échantillons, coefficients de partage; variations dans la performance des dispositifs de purification utilisés, stabilité des réactifs ou des dérivés préparés; effets de la lumière, de la température, des solvants et du stockage sur les analyses dans les extraits; effets des solvants, des injecteurs, des systèmes de séparation sur colonne, des caractéristiques des phases mobiles (composition et vitesse d'écoulement), température, système de détection, co-extractifs, etc. sur le procédé de détermination. Il est très important que les rapports qualitatifs et quantitatifs entre le signal mesuré et l'analyte recherché soient établis sans la moindre équivoque.
4.4.3 On donnera la préférence aux méthodes applicables aux analyses multi-résidus ou multi-matrices. L'emploi d'analytes ou de matrices représentatifs est un outil important pour valider les méthodes. A cette fin, il conviendra de différencier suffisamment les produits, mais pas inutilement. Ainsi, certains produits sont disponibles dans une vaste gamme de variantes manufacturées mineures ou de variétés cultivées, ou de races, etc. En général, mais pas obligatoirement, une variante unique d'un produit particulier peut être considérée comme en représentant d'autres du même produit mais, par exemple, une seule espèce de fruit ou une seule espèce de légume ne doit pas être considérée comme représentant tous les fruits ou tous les légumes (tableau 5). Chaque cas doit être considéré en fonction des avantages qu'il présente, sauf lorsque l'on sait que des variantes dans un produit diffèrent des autres dans leurs effets sur les performances de la méthode. Il peut y avoir de très grandes différences d'une espèce à l'autre dans l'exactitude et la précision des méthodes, en particulier concernant le stade de la détermination.
4.4.3.1 Lorsque l'expérience affiche les mêmes performances en ce qui concerne l'extraction et la purification entre des produits/matrices d'échantillons similaires, une approche simplifiée peut être adoptée pour la validation des performances. Un produit représentatif peut être choisi dans le tableau 5 pour représenter chaque groupe de produits ayant des propriétés communes, et utilisé pour la validation du procédé ou de la méthode. Dans le tableau 5, les produits sont classés selon la Classification Codex[25].
Les données sur la validation d'une méthode peuvent être étendues à d'autres produits de la manière suivante:
4.4.3.2 De la même manière, on peut utiliser des analytes représentatifs pour évaluer les performances d'une méthode. Il faut sélectionner des composés pour couvrir les propriétés physiques et chimiques des analytes devant être déterminés par la méthode. La sélection des analytes représentatifs doit être faite sur la base de l'objectif et du champ d'application de l'analyse en tenant compte des éléments suivants:
(a) Les analytes représentatifs sélectionnés devraient:4.4.3 Lorsque des données appropriées sont déjà disponibles, il n'est pas nécessaire que l'analyste effectue tous les essais. Néanmoins, toutes les informations requises doivent être incluses ou mentionnées dans les registres de validation. Le tableau 1 donne une vue d'ensemble des paramètres à évaluer pour la validation d'une méthode selon le statut de la méthode à valider. Des paramètres et critères spécifiques à évaluer sont énumérés au tableau 2. On évaluera uniquement les paramètres convenant à la fois à la méthode et à l'objectif pour lequel la méthode particulière doit être appliquée. Dans de nombreux cas, les caractéristiques de performance par rapport à plusieurs paramètres peuvent être obtenues simultanément en utilisant une seule expérience. Les plans d'expérience où différents facteurs sont modifiés en même temps (plans expérimentaux factoriels), peuvent aider à réduire au minimum les ressources nécessaires. Les performances de la méthode d'analyse devraient être vérifiées, d'abord durant son élaboration, et ensuite durant son utilisation, comme il est indiqué à la section 4.5, selon les critères mentionnés au tableau 3.(i) Posséder une gamme suffisamment large de propriétés physico-chimiques pour inclure celles des analytes représentés;(b) Dans la mesure du possible, tous les analytes inclus dans le processus de validation initial devraient être ceux qui doivent être testés régulièrement et qui peuvent être déterminés simultanément par le système de dosage utilisé.(ii) Figurer parmi ceux qui ont le plus de chances d'être détectés régulièrement, ou pour lesquels des décisions critiques doivent être prises sur la base des résultats.
(c) La concentration des analytes utilisés pour caractériser une méthode devrait être choisie de manière à couvrir les limites acceptées (LA) de tous les analytes devant être recherchés dans tous les produits. Il s'ensuit que les analytes représentatifs sélectionnés devraient comprendre, entre autres, ceux qui ont des LA élevées et basses. Il s'ensuit que les niveaux d'enrichissement utilisés dans les essais de performance avec des analytes/produits représentatifs pourraient ne pas correspondre aux LA réelles.
4.4.3.1 Les méthodes individuelles (pour un seul résidu) doivent être entièrement validées avec tous les analytes et les produits à analyser spécifiés à cette fin, ou à l'aide de matrices d'échantillon représentatives de celles qui seront testées par le laboratoire.
4.4.3.2 Les méthodes spécifiques de groupe (GSM) devraient être validées au départ avec un ou plusieurs produits représentatifs et un minimum de deux analytes représentatifs choisis dans le groupe.
4.4.3.2 Les méthodes multi-résidus (MRM) peuvent être validées avec des produits représentatifs et des analytes représentatifs.
4.5 Vérification des performances
4.5.1 Les principaux objectifs de la vérification des performances sont les suivants:
Les résultats du contrôle interne de la qualité fournissent des informations essentielles sur la reproductibilité à long terme et d'autres caractéristiques de performance de la méthode, y compris les analytes et les produits qui sont incorporés durant l'extension de la méthode.
Les caractéristiques de performance fondamentales à tester et les procédés appropriés sont décrits au tableau 5.
Pour une vérification efficace des performances, il faut procéder simultanément à l'analyse des échantillons et aux analyses de contrôle de la qualité appropriées (détermination des témoins et des taux de récupération, matériaux de référence, etc.). Afin de vérifier les tendances des performances de la méthode et faire en sorte que le contrôle statistique soit maintenu, on utilisera des diagrammes de contrôle.
4.5.2 Etablissement et utilisation des diagrammes de contrôle.
Le diagramme de contrôle peut être un instrument utile pour démontrer les performances d'une méthode et la reproductibilité du paramètre sélectionné. Exemple, le diagramme de contrôle pour les taux de récupération. Son application sera fonction des tâches du laboratoire. Lorsqu'un grand nombre du même type d'échantillon est analysé pour les mêmes ingrédients actifs, le diagramme de contrôle s'appuie sur le taux de récupération moyen et son-écart type obtenus durant l'utilisation normale de la méthode. Lorsqu'un petit nombre de chaque type d'une grande variété d'échantillons est analysé pour un grand nombre d'analytes avec une procédure multi-résidus, les diagrammes de contrôle peuvent être appliqués de la façon habituelle. Dans de tels cas, au départ, un diagramme de contrôle est établi avec le taux de récupération moyen (Q) des analytes représentatifs dans des matrices représentatives et le coefficient de variation (CVAtyp) de la reproductibilité type en laboratoire, obtenu comme décrit ci-dessous. Lorsque les données sur le taux de récupération moyen et leur coefficient de variation obtenu durant la validation de la méthode pour des analytes/matrices d'échantillon ne sont pas statistiquement différentes, chacune peut être considérée comme une estimation du taux de récupération réel et de la précision de la méthode; en les combinant de façon appropriée, on peut déterminer le taux de récupération type (Qtyp) et le coefficient de variation (CVAtyp) de la méthode et les utiliser pour établir le premier diagramme de contrôle. Les limites d'avertissement et d'action sont Qtyp ± 2*CVAtyp*Q et Qtyp ± 3*CVAtyp*Q, respectivement.
4.5.2.1 Lorsque la méthode est appliquée pour l'analyse normale de diverses combinaisons analyte/matrice représentées durant la validation de la méthode, chaque taux de récupération est indiqué sur le diagramme. La reproductibilité de la méthode durant son utilisation normale pourrait être légèrement supérieure à celle obtenue durant la validation de la méthode. Par conséquent, si certains taux de récupération dépassent occasionnellement les limites d'avertissement, les limites d'action, mais qu'ils sont dans les fourchettes calculées à partir des valeurs CVA spécifiées au tableau 3, il n'y a aucune mesure spéciale à prendre.
4.5.2.2 Sur la base des 15-20 essais de récupération supplémentaires effectués durant l'utilisation normale de la méthode, dans le cadre de la vérification des performances, le taux de récupération moyen ou type et le CVA devront être recalculés et un nouveau diagramme de contrôle sera établi reflétant la reproductibilité à long terme de l'application de la méthode. Les nouveaux paramètres établis doivent se situer dans les fourchettes acceptables spécifiées au tableau 3.
4.5.2.3 Si cela est difficile à réaliser, par exemple dans le cas d'analytes particulièrement problématiques, les résultats obtenus à partir des échantillons devraient être signalés comme présentant une exactitude et une précision moindres que celles qui sont normalement associées à la détermination des résidus de pesticides
4.5.3.4 Durant l'utilisation normale de la méthode, si la moyenne des ³10 premiers essais de récupération pour un analyte/matrice d'échantillon particulier s'écarte sensiblement (P=0,05) du taux de récupération moyen obtenu pour les analytes/matrices d'échantillons représentatifs, les Qtyp et CVtyp ne sont pas applicables. Il faut alors calculer les nouvelles limites d'avertissement et d'action pour l'analyte/matrice d'échantillon particulier, en appliquant le nouveau taux de récupération moyen et les valeurs de coefficient de variation mesurées.
4.5.3.5 Si les données relatives à la vérification des performances dépassent à plusieurs reprises les limites d'avertissement (sur 20 mesures, on peut en accepter une dépassant la limite), il faut vérifier les conditions d'application de la méthode, détecter les sources d'erreur(s) et prendre les mesures correctrices qui s'imposent avant de continuer à utiliser cette méthode.
4.5.3.6 Si les données relatives à la vérification des performances dépassent les limites d'action affinées établies comme au point 4.2.3, il faudra répéter l'essai sur le lot analytique (ou du moins sur les échantillons dans lesquels on trouve des résidus ³ 0,7 LA ou 0,5 LA, pour des analytes détectés régulièrement ou occasionnellement, respectivement).
4.5.3.7 Procéder à une deuxième analyse des portions d'essai d'échantillons positifs pour mieux vérifier les performances. Les résultats peuvent servir à calculer la reproductibilité globale en laboratoire de la méthode (CVLtyp) en général ou pour un analyte/matrice d'échantillon particulier. Dans ce cas, le CVLtyp comprendra aussi l'incertitude du traitement de l'échantillon, mais n'indiquera pas si l'analyte est perdu durant ce processus.
4.6 Epreuves de confirmation
4.6.1 Lorsque les analyses sont effectuées à des fins réglementaires, il est particulièrement important de procéder à des épreuves de confirmation avant de faire un rapport défavorable sur des échantillons contenant des résidus de pesticides qui normalement, ne devraient pas se trouver dans le produit en cause ou si les LMR semblent dépassées. Les échantillons peuvent contenir des substances chimiques perturbatrices qui peuvent être prises à tort pour des pesticides. En chromatographie gazeuse par exemple, on peut mentionner la réponse des détecteurs à capture électronique aux esters de phthalate et celle des détecteurs spécifiques du phosphore aux composés contenant du soufre et de l'azote. Comme première étape, on peut répéter l'analyse en utilisant la même méthode, si une seule portion a été analysée au départ. Cela confirmera la répétibilité du résultat, si le résidu est confirmé. On notera que la seule preuve à l'appui de l'absence de résidus détectables est fournie par les données de vérification des performances.
4.6.2 Les épreuves de confirmation peuvent être quantitatives et/ou qualitatives mais, dans la majorité des cas, il faudra fournir les deux types d'information. Des problèmes particuliers se posent lorsque les résidus doivent être confirmés au seuil de détermination ou à proximité; toutefois, bien qu'il soit difficile de quantifier les résidus à ce niveau, il est essentiel de fournir une confirmation adéquate tant sur le niveau que sur l'identité.
4.6.3 Les épreuves de confirmation nécessaires peuvent être fonction du type d'échantillons et des faits antérieurs connus. Dans un certain nombre de plantes cultivées ou de produits, on trouve presque toujours certains résidus. Dans le cas d'une série d'échantillons de même origine, qui contiennent des résidus du même pesticide, il peut suffire de confirmer l'identité des résidus dans une petite proportion des échantillons choisis au hasard. De manière analogue, lorsque l'on sait qu'un pesticide déterminé a été appliqué au produit échantillonné, la confirmation de l'identité du résidu peut être superflue; il convient néanmoins d'y procéder sur une partie d'échantillons choisis au hasard. Si l'on dispose d'échantillons à blanc, on s'en servira pour déceler la présence éventuelle de substances perturbatrices.
4.6.4 S'il s'agit d'une confirmation qualitative, il est souhaitable de répéter l'épreuve avec au moins une méthode faisant intervenir des propriétés physico-chimiques différentes et/ou des données spectrales. Pour une confirmation quantitative, il faudrait recourir au moins à une autre procédure (qui peut être une technique de détection différente, des ions supplémentaires surveillés par spectrométrie de masse, etc.). Le résultat consigné sera fonction des méthodes appliquées:
Si les deux résultats dépassent les valeurs extrêmes prévues, il faut vérifier la validité de l'un d'eux et consigner la moyenne des deux résultats conformes.
4.6.5 L'analyste doit décider lui-même de la marche à suivre pour identifier un résidu de façon certaine; il s'efforcera tout particulièrement de choisir une méthode permettant d'amoindrir les effets des substances perturbatrices. Dans le choix de la (des) technique(s), il faudra également tenir compte du matériel et des compétences techniques disponibles. D'autres méthodes de confirmation sont présentées au tableau 6.
4.7 Spectrométrie de masse
4.7.1 Les données de résidus obtenues à l'aide de la spectrométrie de masse peuvent représenter la preuve déterminante et, si l'on dispose d'un matériel approprié elle est la technique de choix. Cette technique peut également servir pour le dépistage des résidus. Elle est en général appliquée conjointement avec une technique de séparation chromatographique, ce qui permet d'obtenir des données sur le temps de rétention, le rapport masse/charge des ions et l'abondance de ceux-ci. La technique de séparation particulière, la spectrométrie de masse, l'interface et la gamme de pesticides à analyser sont en général interdépendants et il n'y a pas de combinaison unique se prêtant à l'analyse de tous les composés. La transmission quantitative d'analytes labiles dans le système chromatographique et l'interface pose des problèmes analogues à ceux posés par d'autres détecteurs. La présence d'un résidu est confirmée de façon certaine avec la formation de son spectre complet de masse moyennant ionisation par impact électronique (dans la pratique généralement de m/z50 à un niveau dépassant la région des ions moléculaires). L'abondance relative d'ions dans le spectre et l'absence d'ions pertubateurs jouent un rôle important pour confirmer l'identité. Cette méthode d'analyse est l'une des moins sélectives; il faudrait éviter très soigneusement les perturbations dues à des contaminants introduits durant la production ou le stockage d'extraits. Les systèmes de données de spectrométrie de masse permettent de supprimer les signaux de perturbation de fond (par exemple, perte de colonne) moyennant "soustraction" de ces perturbations, mais il faudra utiliser cette technique avec prudence. On peut en général renforcer la sensibilité par une exploration dans une gamme de masses délimitée ou le contrôle d'ions déterminés, mais plus le nombre d'ions surveillés est petit (particulièrement si leur masse est faible), moins les données obtenues seront définitives. On aura une confirmation supplémentaire de l'identité i) en utilisant la colonne de chromatographie; (ii) en utilisant une autre technique d'ionisation (par exemple ionisation chimique); (iii) en surveillant les autres produits de réaction d'ions déterminés par spectrométrie de masse par tandem (SM/SM ou SMn); ou iv) en surveillant certains ions à une résolution de masse accrue. Concernant la quantification, les ions surveillés devraient être ceux qui sont les plus spécifiques de l'analyte, qui sont sujets à moins de perturbation et fournissent de bons rapports signal-bruit. Les déterminations par la spectrométrie de masse devraient satisfaire aux mêmes critères de contrôle de la qualité de l'analyse que ceux appliqués aux autres systèmes.
4.7.2 La confirmation des résidus détectés après séparation à l'aide de la CLHR pose généralement plus de problèmes que la chromatographie gazeuse. Si la détection se fait par absorption de rayons UV, la production d'un spectre complet peut fournir une bonne preuve de l'identité. Toutefois, les spectres UV de certains pesticides ne sont pas très utiles pour le diagnostic, étant semblables à ceux produits par de nombreux autres composés possédant des groupes ou structures fonctionnels semblables, et la coélution de composés perturbateurs peut créer d'autres problèmes. Les données sur l'absorption d'UV produites à de multiples longueurs d'onde peuvent appuyer ou réfuter l'identification mais, en général, elles ne sont pas suffisamment caractéristiques par elles-mêmes. Les données sur la fluorescence peuvent être utilisées pour appuyer celles obtenues par l'absorption de rayons UV. La CPL-SM peut fournir de bonnes preuves mais, du fait que les spectres produits sont généralement très simples, montrant peu de fragmentation caractéristique, les résultats obtenus avec cette technique n'ont guère de chances d'être définitifs. La CPL-SM/SM est une technique plus efficace, associant sélectivité et spécificité, et qui fournit souvent de bonnes preuves de l'identité d'un pesticide. Les techniques CPL-SM tendent à être sujettes aux effets de matrices, en particulier la suppression, et la confirmation de la quantité pourrait donc exiger le recours à l'addition de produits étalons ou de produits isotypiquement étiquetés. La production de dérivés peut aussi être utilisée pour la confirmation de résidus détectés par la CLHR (paragraphe 4.6.5.4).
4.7.3 Dans certains cas, l'analyse chromatographique en couche mince est le moyen le plus commode de confirmer les résultats de la chromatographie gazeuse. L'identification repose sur deux critères, la valeur de Rf et la réaction de visualisation. Les méthodes de détection fondées sur les titrages biologiques (par exemple, enzymes, spores de champignons, inhibition des chloroplastes) sont particulièrement adaptées à la confirmation qualitative car elles sont spécifiques de certains types de composés, sensibles et normalement très peu affectées par les co-extraits. La documentation scientifique existante contient de nombreuses références à cette technique: L'UICPA Report on Pesticides (13) (Bátora, V., Vitorovic, S.Y., Thier, H.-P. et Klisenko, M.A.; Pure & Appl. Chem., 53, 1039-1049 (1981) fait le point sur cette technique et constitue une bonne introduction. Sur le plan quantitatif toutefois, la chromatographie en couche mince donnent des résultats limités. Un prolongement à cette méthode consiste à retirer la partie de la plaque correspondant au Rf puis à procéder à une élution de la substance à analyser à partir du support, afin de poursuivre la confirmation par analyse chimique ou physique. Il convient toujours de déposer sur la plaque à côté de l'extrait d'échantillon à analyser une tache du pesticide de référence afin d'éviter tout problème de non-répétabilité du fR. Le dépôt d'une tache de pesticide de référence par-dessus la tache d'extrait peut aussi donner des informations utiles. Les avantages de la chromatographie en couche mince sont sa rapidité, son faible coût et son applicabilité à des produits sensibles à l'action de la chaleur. Ses inconvénients sont (en général) une sensibilité et une capacité de séparation inférieures à celles des techniques de détection chromatographiques au moyen d'instruments et le besoin d'une purification plus efficace dans le cas de détections fondées sur des réactions chromatiques des substances chimiques.
4.8 Production de dérivés
Ces méthodes de confirmation peuvent être réparties en trois groupes principaux:
a) Réactions chimiques
On a fréquemment recours à des réactions chimiques de faible ampleur pour obtenir des produits de dégradation, d'addition ou de condensation des pesticides, qui sont ensuite réexaminés par des techniques chromatographiques. Ces produits n'ont ni le même temps de rétention ni les mêmes modalités d'apparition dans les détecteurs que les composés initiaux. Un échantillon de pesticides de référence doit être traité à côté du résidu présumé, de manière à pouvoir comparer directement les résultats. Un extrait enrichi doit également être inclus afin de prouver que la réaction s'est produite en présence d'un échantillon du produit à analyser. Il peut y avoir une perturbation lorsque des produits dérivés sont détectés par les propriétés du réactif dérivé. Un inventaire des réactions chimiques utilisées pour les épreuves de confirmation, a été fait par Cochrane, W.P. (Chemical derivatisation in pesticide analysis, Plenum Press, NY (1981)). Les réactions chimiques ont l'avantage d'être rapides et faciles à effectuer, mais il peut être nécessaire d'acheter des réactifs spéciaux et de les purifier.
b) Réactions physiques
II peut être intéressant de déterminer une altération photochimique d'un résidu de pesticide en vue d'obtenir un ou plusieurs produits ayant un chromatogramme reproductible. Un échantillon de pesticide de référence et un extrait enrichi doivent toujours être traités parallèlement. Si les échantillons contiennent plus d'un seul résidu de pesticides, l'interprétation des résultats peut être difficile. En tels cas, on peut séparer au préalable certains résidus par chromatographie en couche mince, chromatographie en phase liquide à haute résolution ou par fractionnement de la colonne.
c) Autres méthodes
De nombreux pesticides peuvent être dégradés/transformés par des enzymes. Contrairement aux réactions chimiques normales, ces processus sont très spécifiques et entrent généralement dans l'une des catégories suivantes: oxydation, hydrolyse ou de-alkylation. Les produits de conversion ont des caractéristiques chromatographiques différentes du pesticide de départ et la comparaison avec les produits de conversion obtenus avec des pesticides de référence peut être utile pour la confirmation.
4.9 Le concept de concentration étalonnée la plus faible (CEPF)
4.9.1 Lorsque l'analyse a pour objectif de suivre et de vérifier le respect des LMR ou d'autres limites acceptées (LA), les méthodes appliquées aux résidus doivent être suffisamment sensibles pour déterminer de façon fiable les résidus qui pourraient être présents dans une plante cultivée ou dans un échantillon du milieu à ou à proximité de la LMR ou des LA. Toutefois, il n'est pas nécessaire d'utiliser des méthodes ayant une sensibilité permettant de déterminer des quantités de résidus deux ou trois fois inférieures. Les méthodes élaborées pour mesurer les résidus à des concentrations très faibles sont habituellement très chères et difficiles à appliquer. L'emploi des CEPF (voir glossaire) aurait l'avantage de réduire la difficulté technique d'obtenir les données et en outre réduirait les coûts. Les propositions suivantes concernant les CEPF dans divers échantillons pourraient être utiles et permettre aux analystes des résidus de concevoir des méthodes appropriées.
4.9.2 Pour les ingrédients actifs homologués pour lesquels il existe des LMR, les CEPF peuvent être indiquées sous forme de fraction de la LMR. Pour faciliter l'analyse, cette fraction variera et pourrait être comme suit:
|
LMR (mg/kg) |
CEPF (mg/kg) |
|
5 ou plus |
0,5 |
|
0,5 à 5 |
0,1 passant à 0,5 pour des LMR plus
élevées |
|
0,05 à 0,5 |
0,02 passant à 0,1 pour les LMR |
|
moins de 0,05 |
0,5 × LMR |
4.10 Expression des résultats
A des fins réglementaires, seules des données confirmées seront enregistrées, exprimées telles que définies par la LMR. Les valeurs nulles devraient être consignées comme étant inférieures à la concentration étalonnée la plus faible, plutôt qu'inférieures à la concentration calculée par extrapolation. En général, les résultats ne sont pas corrigés en fonction de la récupération, et ils ne peuvent être corrigés que si le taux de récupération s'écarte sensiblement de 100%. Si les résultats sont donnés corrigés en fonction de la récupération, il faut donner à la fois les valeurs mesurées et les valeurs corrigées. Il faut aussi indiquer la base adoptée pour la correction. Lorsque des résultats positifs obtenus par des déterminations répétées (par exemple sur différentes colonnes de CG, avec différents détecteurs ou sur la base d'ions différents des spectres de masse) d'une seule portion d'essai (sous-échantillon), on consignera la valeur la plus basse obtenue. Lorsque des résultats positifs dérivent de l'analyse de plusieurs portions d'essai, on enregistrera la moyenne arithmétique des valeurs les plus faibles obtenues dans chaque portion d'essai. Prenant en compte, en général, une précision relative de 20-30%, les résultats devraient être exprimés seulement avec 2 chiffres significatifs (par exemple: 0,11, 1,1, 11 et 1,1 × 102). Etant donné qu'à de plus faibles concentrations, la précision peut être de l'ordre de 50%, les valeurs de résidus inférieures à 0,1 devraient être exprimées par un chiffre significatif uniquement.
Figure II.1. Tableau synoptique de validation de la méthode
Figure II.2. Vérification de la stabilité de l'analyte
Tableau 1 Résumé des paramètres à évaluer pour la validation des méthodes
|
Paramètres à tester |
Méthode d'analyse existante dont la validité
a déjà été démontrée par des
essais antérieurs du paramètre, pour une ou plusieurs combinaisons
analyte/matrice |
Modification d'une méthode existante |
Nouvelle méthode, pas encore validée |
Types d'expériences pouvant être associés |
||||
|
Vérification des performances* |
Matrice supplémentaire |
Analyte supplémentaire |
Concentration beaucoup plus basse de l'analyte |
Un autre laboratoire |
||||
|
Spécificité (montrer que le signal détecté
est dû à un analyte et non à un composé) |
Non (à condition que critères concernant matrices témoins
et confirmation de observés) |
Oui, si le contrôle de la qualité montre l'interférence
de la matrice |
Oui |
Oui, si le contrôle de la qualité montre l'interférence
de la matrice |
Contrôles rigoureux non nécessaires si la performance du
système de dosage est similaire ou meilleure |
Oui ou non. Des contrôles rigoureux peuvent être nécessaires
si le système de dosage est fondamentalement différent ou
si l'ampleur des interférences de la matrice est |
Oui. Des contrôles rigoureux peuvent être nécessaires
si le système de dosage est différent ou si l'ampleur des
interférences des matrices est incertaine, par rapport aux méthodes
existantes |
|
|
Gamme d'analyse, récupération par extraction, purification,
production de |
Oui |
Oui |
Oui |
Oui |
Oui |
Oui |
Oui |
Gamme d'étalonnage Gamme d'analyse LD/LQ Effet de la matrice |
|
Gamme d'étalonnage pour la détermination de l'analyte |
Non |
Non |
Oui |
Oui |
Oui, pour des analytes |
Oui, pour des analytes |
Oui, pour des analytes représentatifs analytes |
inéarité, reproductibilité et ignal/bruit |
|
LD et LQ |
Non |
Oui, (partiel si la matrice provient d'une classe représentée) |
Oui, partiel pour les analytes représentés |
Oui |
Oui |
Oui |
Oui |
Concentration étalonnée la plus faible et données
de récupération de matériau enrichi à faible
concentration |
|
Limite de notification, CEPF |
Oui |
Non |
Non |
Non |
Non |
Non |
Non |
|
|
Stabilité de l'analyte dans extraits d'échantillon** † |
Non |
Oui, sauf si la matrice provient d'une classe représentée |
Oui, sauf si l'analyte est représenté |
Oui |
Non |
Non, à moins que l'extraction/solvant final soit différente,
ou que la purification soit moins poussée. |
Oui, si l'extraction/solvant final est différente de celle utilisée
dans une méthode existante, ou si la purification est moins poussée
par rapport aux autres méthodes utilisées. |
|
|
Stabilité de l'analyte durant le stockage de l'échantillon**
‡ |
Oui |
Oui |
Oui, |
Théoriquement |
Non |
Non |
Non |
|
|
Efficacité de l'extraction** ¨ |
Non |
Idéalement |
Idéalement |
Théoriquement |
Non |
Non, sauf dans des conditions d'extraction différentes |
Oui sauf si on utilise une procédure d'extraction déjà
testée. |
|
|
Homogénéité** des échantillons à analyser |
Oui* |
Non, sauf si la matrice est sensiblement différente |
Non |
Non |
Non, à moins que l'équipement ne soit changé |
Non, à moins que l'équipement ne soit changé |
Oui, sauf si on utilise un procédé de transformation de
l'échantillon déjà testé |
Voir plus bas |
|
Stabilité de l'analyte dans la transformation de l'échantillon** |
Non |
Oui, à moins qu'il ne s'agisse d'une matrice représentée |
Oui, à moins qu'il ne s'agisse d'un analyte représenté |
Théoriquement |
Non |
Non, à moins que le procédé ne comporte température
plus élevée, laps de temps plus long, broyage plus grossier,
etc. |
Non, à moins que le procédé ne comporte température
plus élevée, laps de temps plus long, broyage plus fin,
etc. que les procédés validés. |
Répétabilité, re-productibilité |
* Contrôle de qualité en cours** Si on ne dispose pas d'informations pertinentes
† II faut choisir des analytes représentatifs sur la base des caractéristiques de l'hydrolyse, de l'oxydation et de la photolyse.
‡ Les données sur la stabilité dans/sur des produits représentatifs devraient fournir des renseignements suffisants. Des essais supplémentaires sont nécessaires, par exemple, lorsque:
|
a |
les échantillons sont stockés pendant un laps de
temps dépassant la période d'essai (par exemple stabilité
testée pendant 4 semaines et qu'une perte de l'analyte mesurable se
produit durant cette période, échantillons non analysés
dans un délai de 6 semaines), |
|
b |
les essais de stabilité sont effectués à
£ -18 ºC, mais que les échantillons
sont stockés en laboratoire à £ 5
ºC; |
|
c |
les échantillons sont normalement stockés
à £ -15 ºC, mais la température
de stockage monte jusqu'à +5 °C). |
¨ L'information sur l'efficacité de l'extraction peut être disponible auprès du fabricant ou de la société qui s'occupe d'homologuer le composé.Tableau 2 Paramètres à évaluer pour la validation de la méthode dans des conditions différentes§ Occasionnellement, avec une analyse répétée de portions d'essai d'échantillons positifs.
|
Paramètre |
Concentration(s) |
Nombre d'analyses ou type d'essai requis |
Méthode quantitative |
Critères |
Observations |
|
|
Méthode de dépistage |
||||||
|
1. Performances de la méthode optimisée en laboratoire
(un seul laboratoire) |
||||||
|
1.1 Stabilité de l'analyte dans les extraits et solutions étalons |
A £ LA, ou avec des résidus bien
détectables |
³ 5 répliques à chaque point approprié dans le temps (y compris zéro) et pour chaque analyte/produit représentatif. Enrichir les extraits de l'échantillon à blanc pour tester la stabilité des résidus. Comparer la concentration de l'analyte dans des solutions étalons
stockées/récemment préparées. |
Aucun changement significatif dans la concentration de l'analyte dans
les extraits et produits étalons à analyser stockés
(P=0,05) |
A la fin de la période de stockage, les résidus ajoutés
à la CEPF sont détectables |
II faudra procéder à un essai de stabilité si la
méthode d'analyse est interrompue durant le procédé
de détermination, et il est probable que le matériau sera
stocké plus longtemps que pour déterminer la précision,
ou si l'on obtient de faibles taux de récupération durant
l'optimisation de la méthode. Si les extraits de récupération
sont stockés durant l'optimisation de la méthode, le taux
de récupération sera mesuré par rapport aux produits
d'étalonnage aussi bien "vieux" que "récemment préparés".
Le temps de stockage devrait comprendre la période la plus longue
qui sera probablement nécessaire pour terminer l'analyse. |
|
|
1.2 Fonction d'étalonnage Effet de la matrice |
CEPF à 2(3) fois la LA |
Tester les fonctions de réponse de tous les analytes inclus dans la méthode avec ³2 répliques à ³3 concentrations de l'analyte plus échantillon à blanc. Pour réponse non linéaire, déterminer la courbe de réponse à ³7 concentrations et ³3 répliques. Tester l'effet de la matrice avec tous les analytes et matrices représentatifs.
Appliquer les produits étalons préparés dans le solvant
et échantillonner les extraits au hasard. |
Pour étalonnage linéaire: coefficient de régression des solutions de produits étalons à analyser ® ³ 0,99. ET de résidus (Sy/x) £0,1 Pour la fonction polynomiale ® ³ 0,98. L'effet de la matrice est confirmé si la différence est
significative pour P = 0,05. |
Pour étalonnage linéaire: coefficient de régression
® ³ 0,98. ET de résidus £
0,2 Pour la fonction polynomiale ® ³ 0,95 |
On peut établir des paramètres d'étalonnage durant l'optimisation du procédé, la détermination de la précision ou la capacité de détection. Préparer des solutions d'étalonnage à des concentrations différentes. Pour les MRM, procéder à l'étalonnage avec des mélanges
d'analytes ("mélange de produits étalons"), qui peuvent
être correctement séparés par le système chromatographique.
Utiliser des produits étalons concordant avec la matrice pour de
nouveaux essais si l'effet de la matrice est significatif. La validation
de la méthode pourrait ne pas donner d'information définitive
sur les effets de la matrice, car ceux-ci changent avec le temps, avec
l'échantillon (parfois), avec la colonne, etc. |
|
|
1.3 Gamme à analyser, exactitude, justesse, précision,
limite de détection (LD), limite de quantification (LQ) |
CEPF à 2 (3) fois la LA* |
Analyser combinaisons matrices/analytes représentatifs: ³
5 portions à analyser enrichies à zéro, CEPF, LA
et ³ 3 répliques à des concentrations
de 2-3 LA. Les essais de récupération devraient être
partagés entre tous les analystes qui utiliseront la méthode
et les instruments qui serviront pour l'analyse. |
La LQ devrait convenir à l'objectif. Taux de récupération
moyen et CVA voir tableau 2. Valeur moyenne des résidus*
mesurée dans le matériau de référence ne diffère
pas sensiblement de la valeur convenue (P = 0,05). |
Toutes les récupérations sont détectables à
la CEPF |
Les analystes devraient démontrer que la méthode convient pour déterminer la présence de l'analyte à la LA appropriée, avec le maximum d'erreurs (faux négatifs et faux positifs) spécifié. Pour les MRM, le niveau d'enrichissement des échantillons à blanc devrait comprendre les LA des analytes représentés. Par conséquent, celles-ci pourraient ne pas correspondre à la LA effective pour les analytes représentatifs. Enrichir les portions à analyser avec des mélanges de substances étalons. Les gammes d'exactitude et de précision déterminées pour les combinaisons analyte/matrice représentatives peuvent être considérées typiques de la méthode, et seront utilisées comme critères d'applicabilité pour l'extension à de nouveaux analytes et produits, et premières orientations pour le contrôle interne de la qualité de la méthode. Consigner les résultats non corrigés, le taux de récupération moyen et le CVA des répliques. Le CVA équivaut à la reproductibilité en laboratoire de l'analyse des échantillons. * Corriger les résultats pour le taux de récupération moyen si celui-ci est très différent de 100 %. Lorsque la méthode ne permet pas d'estimer le taux de récupération,
l'exactitude et la précision seront celles de l'étalonnage. |
|
|
1.4 Spécificité et sélectivité de la détection
de l'analyte |
A la concentration étalonnée la plus faible (CEPF) |
Identifier par la spectrométrie de masse, par une technique similaire ou en combinant de façon appropriée les techniques de séparation et de détection disponibles. Analyser ³ 5 blancs de chaque produit représentatif, obtenus de préférence auprès de sources différentes. Consigner l'équivalent de l'analyte dans la réponse du blanc. Déterminer et consigner la sélectivité (d)
du détecteur et les facteurs de réponse relatifs (fRR) des
analytes représentatifs avec les détecteurs spécifiques
utilisés. |
La réponse mesurée est due uniquement à l'analyte. Les résidus mesurés sur deux colonnes différentes
devraient être compris dans la gamme critique des déterminations
chromatographiques repétées. |
Le taux d'échantillons avec de faux résultats négatifs
(erreur b) à la LA devrait être
en général < 5%. |
S'applique uniquement à une combinaison particulière de techniques de séparation et de détection. Au lieu d'échantillons non traités, on pourra utiliser des échantillons qui ont été soumis à des traitements que l'on connaît, pour des analytes autres que ceux appliqués durant le traitement. La maturité des matrices de l'échantillon peut influer dans une large mesure sur la réponse de l'échantillon à blanc. Il faudra contrôler régulièrement les valeurs du blanc durant la vérification des performances (voir Section 5 ci-dessous). Consigner les pics typiques présents dans les extraits d'échantillons à blanc. La CEPF devrait de préférence être £ 0,3 LA, sauf lorsque la LA est fixée à la limite de quantification ou à proximité. L'essai peut être effectué en même temps que la détermination de la limite de décision et la capacité de détection et fournira aussi des informations sur les tRR et fRR des composés. Modifier les conditions chromatographiques si l'échantillon à
blanc influe sur l'analyte ou utiliser un autre système de détection.
La combinaison appropriée de détecteurs sélectifs
augmente la spécificité, car la quantité d'informations
sur l'analyte augmente. |
|
|
1.5 Sélectivité de la séparation |
A la LA |
Déterminer les valeurs de tRR pour tous les analytes à
tester par la méthode (pas seulement les composés de référence).
Lorsqu'on utilise des techniques chromatographiques sans détection
spectrométrique, il faut appliquer différents principes
de séparation et/ou déterminer les tRR sur des colonnes
à polarité différente. Déterminer et consigner
la résolution (RS) et les facteurs de traîne (Tf)
des pics critiques. |
Le pic maximal le plus proche devrait être séparé
du pic désigné de l'analyte par au moins une largeur entière
à 10% de la hauteur du pic; sinon, une détection plus sélective
de tous les analytes est nécessaire. |
Identification provisoire de tous les analytes testés (il n'est
pas nécessaire de séparer tous les analytes) |
A moins d'utiliser la séparation chromatographique et la détection spectrométrique en combinaison, consigner les valeurs de tRR sur des colonnes de polarité différente, qui permettent de séparer (minimum R ³ 1,2) tous les analytes testés. L'essai peut être associé avec la détermination de
la fonction d'étalonnage et l'effet de la matrice (voir 1.7) |
|
|
1.6 Homogénéité de l'analyte dans l'échantillon
à analyser |
A proximité de la limite acceptée ou résidus bien
détectables |
Analyser ³5 portions identiques d'un échantillon d'essai d'un produit représentatif de chaque groupe (Tableau 4), après traitement. Déterminer le CVSp avec l'analyse de la variance. On vérifiera l'homogénéité de l'analyte avec
des analytes dont la stabilité a été reconnue. |
CVSp £ 10%. |
CVSp £ 15% Pour les méthodes de dépistage, il est parfois préférable
de prélever une portion dans laquelle on peut prévoir qu'il
y aura plus de résidus (par exemple peau d'agrume) et il n'est
pas toujours nécessaire qu'elle soit homogène. |
Utiliser de préférence des produits dont les résidus d'origine à la surface sont stables ou traiter la surface d'une petite partie des unités naturelles (<20%) de l'échantillon de laboratoire avant de couper ou de hacher pour représenter le scénario le plus défavorable pour le traitement de l'échantillon. Traitement validé pour l'utilisation avec tout procédé ultérieur. Validation applicable à d'autres produits ayant les mêmes propriétés physiques et indépendante de l'analyte. On peut en même temps tester la stabilité de l'analyte (voir Section 1.7 de ce tableau) Déterminer la constante d'échantillonnage[26],[27]. pour calculer la taille de la portion à analyser requise pour satisfaire aux critères de qualité du CVSp < 10% spécifié. Il peut ne pas être nécessaire de déterminer séparément
le CVSp si le CVL des résidus d'origine se
situe dans les limites spécifiées au tableau 2. |
|
|
1.7 Stabilité de l'analyte durant le traitement de l'échantillon |
A proximité de la LA |
Enrichir les produits avec des quantités connues d'analytes avant de traiter l'échantillon. Analyser ³ 5 répliques de chaque produit, après traitement, Appliquer un composé marqueur théoriquement stable avec
les analytes soumis à l'essai. Pour les MRM et les méthodes
pour des groupes spécifiques, il est possible de tester ensemble
plusieurs analytes. |
Il n'est pas nécessaire de spécifier la stabilité de l'analyte si le taux de récupération global moyen de l'analyte ajouté avant le traitement de l'échantillon (y compris la récupération du procédé) et le CVA se situent dans les fourchettes indiquées au tableau 2. Quantifier la stabilité si la récupération globale
et la récupération du procédé diffèrent
sensiblement (P=0,05). |
L'analyte ajouté à la CEPF reste détectable après
le traitement |
La température de l'échantillon durant le traitement peut être critique. Traitement validé pour être utilisé (...) procédé ultérieur. La validation peut être spécifique (...) et/ou de la matrice de l'échantillon. Pour tester la stabilité, déterminer le taux de récupération moyen et le CVL des composés marqu(...)bles. Utiliser ces composés pour des contrôle (...) qualité (voir section 5). Exprimer le ratio de la concentration moyenne des composés labiles
et stables pour indiquer la stabilité des résidus. Les CV
des composés stables indiqueront également la répétibilité
en laboratoire. |
|
|
1.8 Efficacité de l'extraction |
A proximité de la LA ou résidus facilement mesurables |
Analyser ³5 portions identiques d'échantillons ou de matériau de référence avec des résidus d'origine. Comparer le procédé de référence (ou différent) avec celui faisant l'objet de l'essai. Pour les MRM, il serait préférable que les analytes testés
aient une large gamme de coefficients de partage octanol/eau. On les déterminera
uniquement en utilisant les résidus d'origine. |
Pour les échantillons contenant des résidus d'origine, le résultat moyen obtenu avec le procédé de référence et le procédé à l'essai ne devrait pas différer sensiblement de la concentration P=0,05 en appliquant CVL dans le calcul. Ou, la valeur convenue du matériau de référence et la moyenne des résidus ne devraient pas différer sensiblement à une concentration de P=0,05 lorsqu'elles sont calculées avec le CVA de la méthode testée. Lorsque le CVA de la méthode dépasse 10%, il faudrait augmenter le nombre d'analyses répétées pour maintenir l'écart-type relatif de la moyenne < 5%. Sinon, quantifier et consigner l'efficacité de l'extraction (non
compris la récupération de la phase analytique après
l'extraction). |
La moyenne de résidus d'origine, qui sont présents à
ou à proximité de la LQ ou de la CEPF, est détectable
dans les échantillons. |
La température de l'extrait, la vitesse du mélangeur ou Ultra Turrax, la durée de l'extraction et le rapport solvant/eau/matrice peuvent avoir un effet sensible sur l'efficacité de l'extraction. On peut vérifier l'effet de ces paramètres en effectuant un essai de robustesse. Les conditions optimisées devraient être maintenues constantes autant que possible. La validation est généralement applicable aux produits appartenant à un même groupe et aux analytes représentés ayant des propriétés physiques et chimiques similaires. La validation est indépendante des procédés qui seront appliqués ultérieurement. Le taux de récupération moyen de chaque méthode devra être déterminé sur des portions à analyser enrichies. Corriger les résultats avec un taux de récupération moyen de l'analyse s'il est très différent de 100%. Selon certains règlements, la capacité des nécessaires
de dépistage doit être testée pour détecter
un résultat positif avec un taux de confiance de 95%. |
|
|
1.9 Stabilité de l'analyte durant le stockage de l'échantillon |
A proximité de la limite acceptée |
Analyser des échantillons venant d'être homogénéisés contenant des résidus d'origine, ou homogénéiser et enrichir des échantillons à blanc (temps 0). puis analyser les échantillons stockés selon les procédés normaux du laboratoire (en général à £ -18 ºC). La durée du stockage devrait être ³ à l'intervalle le plus long prévu entre l'échantillonnage et l'analyse. ³ 5 répliques à chaque point
dans le temps. Quand les portions stockées sont analysées
³ 4 occasions, tester ³ 2 portions enrichies, et ³ 1 portion témoin enrichie au moment de l'analyse. |
Aucune perte significative de l'analyte durant le stockage (P = 0,05) |
L'analyte ajouté à la concentration étalonnée
la plus faible (CEPF) reste détectable après le stokage |
L'entreposage est validé pour l'emploi avec tout procédé ultérieur. La validation est propre à l'analyte. Toutefois, en général, les données sur la stabilité à l'entreposage obtenues avec des matrices d'échantillons représentatifs peuvent être considérées valides pour des matrices semblables. Les matrices devront être choisies compte tenu de la stabilité chimique (par exemple hydrolyse) de l'analyte et l'utilisation prévue de la substance. L'information utile sur la stabilité durant le stockage peut être obtenue avec les évaluations de la JMPR[28] ou dans la documentation présentée pour l'enregistrement des composés. Consigner la concentration initiale de résidu, la concentration du résidu restante et la récupération de l'analyte dans le procédé. On évitera de stocker inutilement l'échantillon en planifiant
(...)sement l'échantillonnage et l'analyse consécutive,
(...)nant un arrangement administratif qui ne fait pas partie de la méthode
d'analyse. |
|
|
2. Extension de la méthode validée |
||||||
|
2.1 Stabilité de l'analyte durant le stockage et la transformation
des échantillons, et dans les extraits et solutions de substances
étalons. |
Voir 1.1, 1.2 et 1.9 |
|
|
|
Seulement si on ne dispose pas encore de données sur la stabilité
dans les conditions de transformation et sur la matrice représentative. |
|
|
2.2 Fonction d'étalonnages et de la matrice |
CEPF à 2 (3) fois la LA: |
Etalonnages en trois points qui comprennent la limite acceptée
avec et sans substances étalons à analyser concordant avec
la matrice |
Pour l'étalonnage linéaire: coefficient de régression pour les solutions de substances étalons à analyser ® ³ 0,99. ET des résidus relatifs (Sy/x) £ 0,1 Pour la fonction polynomiale ® ³ 0,98. |
Pour l'étalonnage linéaire: coefficient de régression ® ³ 0,98. ET des résidus relatifs £ 0,2 Pour la fonction polynomiale ® ³ 0,95. |
La validation de la méthode peut ne pas donner d'informations
précises sur les effets de la matrice, du fait que ceux-ci changent
dans le temps, avec l'échantillon (parfois), avec la colonne, etc. |
|
|
2.3 Exactitude, précision, LD, LQ |
A la LA |
Dans le cas d'une détection prévue: a) Analyser 3 portions de matrices d'échantillons représentatifs intéressant l'analyste enrichis à la limite acceptée. Dans le cas d'une détection imprévue: b) Enrichir deux ou, mieux, trois portions supplémentaires de
l'échantillon à analyser à peu près à
la concentration du nouvel analyte. Calculer le taux de récupération
de l'analyte ajouté. Utiliser une matrice d'échantillon
similaire pour l'essai de récupération si on ne dispose
pas d'une quantité suffisante d'échantillon à analyser. |
Les résidus récupérés devraient se situer dans les limites de répétabilité de la méthode: Trois portions: Cmax - Cmin £ 3.3CVAtyQ Deux portions: Cmax - Cmin £ 2,8*CVAtypQ CVAtyp est le coefficient de répétabilité type de variation de la méthode à adapter. Q = récupération moyenne du nouvel analyte; il doit être
conforme au tableau 2. |
Les analytes ajoutés aux échantillons à blanc à
la concentration visée indiquée devraient être mesurables
dans tous les essais. |
Utiliser le CVAtyp établi durant la validation de la
méthode. La méthode ne devrait être testée
qu'avec des produits représentant l'utilisation prévue (mauvaise
utilisation possible) de l'analyte. |
|
|
2.4 Spécificité et sélectivité de la détection
de l'analyte |
A la CEPF |
Identifier par spectrométrie de masse, ou en combinant de façon appropriée les techniques de séparation et de détection disponibles. Dans le cas d'une détection prévue: a) Analyser un échantillon à blanc représentatif de chaque groupe de produits intéressant l'analyste (dans lesquels le nouvel analyte sera probablement présent). Analyser une nouvelle matrice avec des composés représentatifs. Dans le cas d'une détection imprévue: b) Vérifier la réponse de l'échantillon à blanc (si disponible), ou démontrer que la réponse obtenue correspond seulement à l'analyte. en utilisant la meilleure technique disponible au laboratoire. Vérifier d et la fRR de détection
ainsi que les tRR des analytes représentatifs. Comparer les tRR
et la réponse du nouvel analyte avec d'autres analytes testés
durant la validation de la méthode et avec les réponses
des blancs obtenues durant l'extension de la méthode et la validation
antérieure de la méthode. |
La réponse mesurée est due uniquement à l'analyte. Le système de détection utilisé devrait afficher une performance du détecteur égale ou supérieure à celle appliquée durant la validation de la méthode. Les résidus mesurés sur deux colonnes différentes
devraient se situer dans la fourchette critique des déterminations
par chromatographie des répliques. Les tRR des analytes représentatifs
obtenus durant la validation de la méthode ne devraient pas dépasser
2 % pour la détermination par CPG et 5 % pour la détermination
par CLHR. |
Le taux d'échantillons donnant de faux résultats négatifs
(erreur b) à la limite acceptée
devrait être < 5%. |
Lorsque l'on prévoit d'étendre la méthode à un nouvel analyte, il faut vérifier l'applicabilité de la méthode pour toutes les matrices d'échantillons représentatifs dans lesquelles l'analyte peut être présent. Lorsqu'un analyte est détecté de façon imprévue, le contrôle de la performance peut être effectué pour la matrice réelle seulement Voir aussi 1.4. Les réponses des échantillons à blanc ne doivent pas interférer avec les analytes, qui seront probablement mesurés dans l'échantillon. Consigner les pics typiques présents dans les extraits d'échantillons à blanc. Le bruit de fond d'un extrait d'une nouvelle matrice doit être compris dans la fourchette obtenue pour les produits/matrices d'échantillon représentatifs. Si la sélectivité de la détection n'élimine
pas la réponse de la matrice, utiliser une combinaison appropriée
de colonnes de chromatographie qui permet de séparer les analytes
des pics de la matrice. Voir d'autres options au tableau 3. |
|
|
2.5 Sélectivité de la séparation |
Voir 1.5 |
Voir 1.5 |
Voir 1.5 |
Voir 1.5 |
Voir 1.5 Seulement si on ne dispose pas d'informations |
|
|
2.6 Efficacité de l'extraction |
Voir 1.8 |
Voir 1.8 |
Voir 1.8 |
Voir 1.8 |
Voir 1.8 Seulement si on ne dispose pas d'informations |
|
|
3. Adaptation de la méthode validée dans un autre laboratoire |
||||||
|
3.1 Pureté et adéquation des substances chimiques, réactifs
et ad(ab)sorbants |
|
Tester un blanc de réactif et l'applicabilité des ad(ab)sorbants
et réactifs. Produire des dérivés avec et sans échantillon. |
Aucune réponse d'interférence dépassant 0,3 CEPF. |
Aucune réponse d'interférence dépassant 0,5 CEPF. |
Quelques-uns des problèmes les plus communs dans le transfert
des méthodes concernant des différences dans le choix des
réactifs, des solvants, des milieux de chromatographie ou dans
les dotations en équipement. Chaque fois que possible, essayer
de confirmer les matériels et l'équipement utilisés
par le concepteur de la méthode, si cette information n'est pas
fournie avec la méthode ou la publication reçue. Une fois
que la méthode fonctionne dans le laboratoire, on peut essayer
d'effectuer des substitutions. |
|
|
3.2 Stabilité de l'analyte dans des extraits et solutions de substances
étalons |
Voir 1.10 |
Voir 1.1 |
Voir 1.1 |
Voir 1.1 |
Cet essai peut être omis si des informations complètes sont
fournies avec la méthode sur la stabilité de l'analyte ou
si la méthode en remplace une autre utilisée précédemment
pour l'analyte, et que des données sur la stabilité de l'analyte
ont déjà été fournies pour la méthode
précédente. |
|
|
3.3 Fonction d'étalonnage Effet de la matrice |
CEPF à 2(3) fois la LA |
Tester les fonctions de réponse des analytes représentatifs inclus dans la méthode à ³ 3 concentrations d'analytes plus le blanc. Pour une réponse non linéaire, déterminer la courbe de réponse à ³ 7 concentrations et ³ 3 répliques. Tester l'effet de la matrice avec des analytes et matrices représentatifs. |
Pour l'étalonnage linéaire: coefficient de régression pour des solutions de substances étalons à analyser ® ³ 0,99. ET des résidus relatifs (Sy/x) £ 0,1 Pour la fonction polynomiale ® ³ 0,98. |
Pour l'étalonnage linéaire: coefficient de régression ® ³ 0,98. ET des résidus relatifs £ 0,2 Pour la fonction polynomiale ® ³ 0,95. |
Voir: 1.2 |
|
|
3.4 Gamme d'analyse Exactitude et précision, limite de détection,
limite de quantification |
Extrait en blanc et/ou à la LA |
Analyser des combinaisons analyte/matrice représentatives: ³ 5 portions à analyser de chaque échantillon à blanc enrichi à 0 et à la LA, et 3 portions enrichies à 2 LA. Les essais de récupération doivent être répartis
entre tous les analystes qui utiliseront la méthode et les instruments
qui serviront à l'analyse. |
La récupération moyenne et le CVA doivent être
compris dans les fourchettes indiquées au tableau 2. |
Toutes les récupérations seront détectables à la CEPF. Matériels de référence à la LA: analyte détecté. |
Voir observations à la section 1.3. |
|
|
3.5 Spécificité et sélectivité de la détection
de l'analyte |
A la LA |
Vérifier les caractéristiques de performance des détecteurs
utilisés et les comparer avec celles spécifiées dans
la méthode. Vérifier la réponse d'un blanc de chaque
produit représentatif, ou effectuer l'essai selon la description
à la section 1.4. |
La réponse mesurée est due uniquement à l'analyte.
Les performances du détecteur (sensibilité et sélectivité)
doivent être égales ou supérieures à celles
spécifiées dans la méthode. Voir section 1.4 |
Le taux d'échantillons donnant de faux résultats négatifs
(erreur (ß) à la LA doit être en général
< 5%. |
La réponse relative des détecteurs spécifiques peut varier sensiblement d'un modèle à l'autre. Une vérification correcte de la spécificité de la détection est critique pour obtenir des résultats fiables. Comparer la réponse du blanc observée avec les pics typiques
signalés dans les extraits en blanc. Voir d'autres observations
à la section 1.4. |
|
|
3.6 "Homogénéité" de l'analyte |
A proximité de la LA ou résidus bien détectables |
Tester deux produits représentatifs de nature différente |
CVSp<10% |
CVSp<15% Pour les méthodes de dépistage, il est parfois préférable
de prélever une portion dans laquelle on peut prévoir qu'il
y aura plus de résidus (par exemple peau d'agrume) et il n'est
pas toujours nécessaire qu'elle soit homogène. |
Les essais sont menés pour confirmer la similitude des conditions
d'application et l'applicabilité des paramètres obtenus
par le laboratoire validant la méthode. Quand l'essai donne des
résultats indiquant un CVSp similaire, les conditions
de transformation de l'échantillon peuvent être considérées
analogues et il ne sera pas nécessaire de procéder à
d'autres essais pour la validation de la méthode. |
|
|
3.7 Stabilité de l'analyte dans les extraits et solutions étalons |
Voir 1.1 |
Voir 1.1 |
Voir 1.1 |
Voir 1.1 |
Cet essai peut être omis si des informations complètes sont
fournies avec la méthode sur la stabilité de l'analyte ou
si la méthode en remplace une autre utilisée précédemment
pour l'analyte, et que des données sur la stabilité de l'analyte
ont déjà été fournies pour la méthode
précédente. |
|
|
Concentration |
Répétibilité |
Reproductibilité |
Justesse2 |
||
|
CVA%3 |
CVL%4 |
CVA%3 |
CVL%4 |
Gamme de taux moyens de récupération |
|
|
£ 1 µg/kg |
35 |
36 |
53 |
54 |
50-120 |
|
> 1 µg/kg £ 0,01
mg/kg |
30 |
32 |
45 |
46 |
60-120 |
|
> 0,01 mg/kg £ 0,1 mg/kg |
20 |
22 |
32 |
34 |
70-120 |
|
>0,1 mg/kg £ 1 mg/kg |
15 |
18 |
23 |
25 |
70-110 |
|
> 1 mg/kg |
10 |
14 |
16 |
19 |
70-110 |
1. Avec les méthodes multi-résidus, il peut y avoir des analytes dans lesquels ces critères de performance quantitatifs ne peuvent pas être strictement observés. L'acceptabilité des données produites dans ces conditions dépendra du but des analyses, par exemple lorsque l'on vérifie la conformité aux LMR, les critères indiqués devraient être observés dans la mesure où cela est techniquement possible, tandis que toute donnée au-dessous de la LMR pourrait être acceptable avec la plus grande incertitude.Tableau 4 Spécifications pour la vérification des performances2. Ces gammes de taux de récupération sont appropriées pour les méthodes multi-résidus. Des critères plus stricts peuvent être nécessaires dans certains buts, par exemple méthodes pour des analytes uniques ou des résidus de médicaments vétérinaires (voir Codex Volume 3, 1996).
3. CVA: Coefficient de variation pour l'analyse excluant la transformation de l'échantillon. Le paramètre peut être estimé à l'aide d'essais effectués avec des matériaux de référence ou des portions à analyser enrichies avant l'extraction. En l'absence d'un matériel de référence certifié, on peut utiliser un matériel de référence préparé au laboratoire.
4. CVL: C'est le coefficient de variation global d'un résultat de laboratoire, prévoyant jusqu'à 10% de variabilité dans la transformation de l'échantillon.
|
Paramètre |
Concentration(s) |
Nombre d'analyses ou type d'essai requis |
Méthode quantitative |
Critère |
Observations |
||
|
Méthode de dépistage |
|||||||
|
4. Contrôle de la qualité (vérification des performances) |
|||||||
|
4.1 Méthodes utilisées régulièrement |
|||||||
|
4.1.1 Adéquation des substances chimiques, adsorbants et réactifs |
|
Pour chaque nouveau lot: tester un blanc de réactifs, l'applicabilité
des ad(ab)sorbants et des réactifs.Procéder à la
production de dérivés sans échantillon. |
Pas de réponse d'interférence (0,3 CEPF. |
Pas de réponse d'interférence (0,5 LA. |
Autrement, si l'échantillon à blanc, l'étalonnage
et la récupération sont satisfaisants, l'adéquation
des réactifs, etc. est confirmée. |
||
|
4.1.2 Etalonnage et gamme de l'analyse |
|
L'étalonnage en un point unique peut être utilisé
avec des mélanges de substances-étalons, si l'intersection
de la fonction d'étalonnage est proche de 0.Appliquer l'étalonnage
en des points multiples (3×2) pour la confirmation quantitative. |
Le lot à analyser peut être considéré comme
étant sous contrôle statistique si les substances-étalons
à analyser et les extraits d'échantillon sont injectés
alternativement, et l'écart-type calculé des résidus
relatifs est (0,1. |
L'analyte est détecté à la CEPF. |
La solution étalon et les échantillons doivent être
injectés alternativement.L'échelonnement avec des injections
de substances-étalons appropriées peut constituer une alternative
à l'étalonnage multi-points qui fera gagner du temps, en
particulier si on ne dispose pas d'un échantillonneur automatique.Etant
donné que la réponse du système change souvent, on
peut procéder à un étalonnage multi-points régulièrement
pour confirmer que l'intersection est proche de zéro.L'étalonnage
multi-points n'est pas nécessaire pour la confirmation quantitative
si le produit étalonné a une concentration très proche
de celle de l'échantillon. |
||
|
4.1.3 Exactitude et précision |
Dans la gamme de l'analyse |
Inclure dans chaque lot à analyser (1 échantillon enrichi
avec un mélange de substances étalons, ou effectuer une
nouvelle analyse d'une portion réplique d'un échantillon
positif, |
La performance du détecteur et la colonne de chromatographie seront
égales ou supérieures à celles spécifiées
dans la méthode.Il est préférable que tous les taux
de récupération restent dans la limite d'avertissement du
diagramme de contrôle établi selon la section 4.2. Pour un
essai long, un sur 20 ou 100 échantillons peut dépasser
les limites d'avertissement et d'action, respectivement. Le lot à
analyser devrait être répété si un des taux
de récupération s'inscrit en dehors des limites d'action,
ou si les résultats des analyses répétées
de l'échantillon positif dépassent la gamme critique.Cmax
- Cmin > 2.8*CVLtypQQ est le résidu moyen
obtenu à partir des mesures répétées, le CVLtyp
est la mesure de la reproductibilité en laboratoire qui comprend
l'incertitude combinée de la transformation et de l'analyse de
l'échantillon. |
Enrichir la portion à analyser avec un ou plusieurs mélanges
de substances-étalons. Modifier ces mélanges en différents
lots afin d'obtenir des taux de récupération pour tous les
analytes intéressant l'analyste à intervalles réguliers.
Effectuer alternativement des études de récupération
à la LA ainsi qu'à la CEPF et à 2 fois la LA, selon
le cas, pour confirmer l'applicabilité de la méthode dans
la gamme d'analyse. Les études de récupération à
la LA devront être deux ou trois fois plus fréquentes que
celles menées à d'autres niveaux.L'analyse répétée
d'échantillons positifs peut remplacer l'essai de récupération
dans un lot particulier.Pour les MRM, préparer des mélanges
de substances étalons spécifiques du produit échantillon
provenant des analytes pouvant se trouver dans un échantillon particulier.
La sélection des analytes pour un mélange devrait assurer
la séparation/détection sélective sans poser de problème.Pour
une identification provisoire: préparer des lots à analyser
contenant le mélange approprié pour l'essai de détection
et les échantillons.Pour la détermination/confirmation quantitative,
inclure dans le lot à analyser le mélange d'essai de détection,
un nombre approprié de mélanges d'étalonnage, un
ou plusieurs échantillons à blanc enrichis, ou un échantillon
réplique positif et les nouveaux échantillons positifs.
Injecter alternativement substances-étalons et échantillons. |
|||
|
4.1.4 Sélectivité de la séparation, spécificité
de la détection, performance des détecteurs |
|
Inclure un mélange d'essai de détection approprié
dans chaque lot de chromatographie. Inclure un produit non traité
(si disponible) dans le lot à analyser. Ajouter des substances
étalons si aucun échantillon non traité est disponible
(semblables à celles analysées dans le lot)Confirmer l'identité
et la quantité de chaque analyte présent à (0,7 de
la LA. |
Les valeurs Rs, Tf des composés soumis à
l'essai, et fRR et (de la détection devraient être comprises
dans la fourchette indiquée.Les tRR ne devraient pas dépasser
2 % pour la détermination avec la CGL et 5 % pour la détermination
avec la CLHR. La performance du détecteur devrait être comprise
dans les limites spécifiées. Les co-extractifs d'échantillon
interférant avec l'analyte ne devraient pas être présents
à (0,3 CEPF. La récupération de la substance-étalon
ajoutée devrait être comprise dans une gamme de récupération
acceptable de l'analyte. |
La performance du détecteur doit être comprise dans les
limites spécifiées. L'analyte doit être > CEPF
ou CC(pour les composés interdits. |
Appelé aussi parfois essai d'"adéquation du système".
Préparer un mélange pour l'essai de détection pour
chaque méthode de détection. Sélectionner les éléments
du mélange afin d'indiquer les paramètres caractéristiques
de la séparation chromatographique et de la détection. Adapter
la base de données sur la rétention relative pour les composés
du mélange d'essai de détection et les analytes utilisés
pour l'étalonnage. Définir la valeur de fRR spécifique
pour le système de détection. Procéder à une
confirmation quantitative avec des substances étalons préparées
dans un extrait de matrice en blanc si l'effet de la matrice est important. |
||
|
4.1.5 Homogénéité de l'analyte dans l'échantillon
traité |
A une concentration de l'analyte facile à détecter. |
Choisir au hasard un échantillon positif. Répéter
l'analyse d'une ou deux autres portions à analyser. |
Les résidus mesurés deux jour différents doivent
être compris dans la limite de reproductibilité des portions
répliques à analyser:Cmax - Cmin (2.8*CVLtypQQ
est la moyenne de résidu obtenue à part mesures des répliques,
CVLtyp est l'incertitude combinée du traitement et de
l'analyse de l'échantillon obtenue durant la validation de la méthode. |
Effectuer les essais alternativement de manière à couvrir
chaque produit analysé. Tester l'homogénéité
au début de la période de croissance ou au début
de l'analyse du type donné d'échantillons.Les résultats
acceptables de l'essai confirment également que la reproductibilité
des analyses (CVA) était appropriée. |
|||
|
4.1.6 Efficacité de l'extraction |
|
|
|
|
L'efficacité de l'extraction ne peut être contrôlée
durant l'analyse. Pour garantir une efficacité appropriée,
la procédure d'extraction validée devrait être effectuée
sans aucun changement. |
||
|
4.1.7 Durée de l'analyse |
|
|
Les échantillons, extraits, etc. ne doivent pas être stockés
pendant un laps de temps dépassant la période pour laquelle
la stabilité au stockage a été testée durant
la validation de la méthode. Les conditions de stockage doivent
être régulièrement contrôlées et consignées. |
Des exemples concernant le besoin d'essais de stabilité supplémentaires
au stockage figurent au tableau 2. |
|||
|
4.2 Analyte détecté occasionnellement |
|||||||
|
EFFECTUER LES ESSAIS DÉCRITS EN 4.1 AVEC LES EXCEPTIONS CI-APRÈS |
|||||||
|
4.2.1 Exactitude et précision |
A proximité de la LA |
Analyser une autre portion d'essai;Ajouter des substances étalons
à la concentration de l'analyte mesurée. |
Les résidus mesurés deux jours différents doivent
être compris dans la fourchette critique:Cmax - Cmin (2.8*CVLtypQQ
est la moyenne de résidu obtenu à partir des mesures répétées,
tandis que le CVLtyp est obtenue durant la validation de la
méthode.Le taux de récupération après l'addition
de substances-étalons doit être compris dans les limites
d'action. |
Vérifier l'exactitude si on trouve un résidu à (0,5
LA. |
|||
|
4.3 Méthodes utilisées à intervalles irréguliers |
|||||||
|
Effectuer les essais décrits en 4.1 avec les exceptions ci-après |
|||||||
|
4.3.1 Exactitude et précision (répétibilité) |
A la LA et à la CEPF |
Inclure un échantillon enrichi à la CEPF et deux échantillons
à la LA dans chaque lot à analyser.Ajouter des substances
étalons si on ne dispose pas d'un échantillon non traité
(semblable à celles utilisées dans le lot).Procéder
à l'analyse avec (2 portions à analyser. |
Au minimum deux taux de récupération doivent être
dans la limite d'avertissement et un dans la limite d'action.Les résidus
mesurés dans les portions répliques doivent être compris
dans la fourchette critique:Cmax - Cmin < 2.8*CVLtypQ
or Cmax - Cmin (f(n) * CVLtypQQ
est la moyenne de résidu obtenue à partir des mesures répétées,
tandis que le CVLtyp est obtenu durant la méthode de
validation, f(n) est le facteur pour le calcul des valeurs
extrêmes qui est fonction du nombre d'échantillons répliques. |
Les résultats acceptables démontrent également l'adéquation
des substances chimiques, adsorbants et réactifs utilisés.Confirmer
les résidus dépassant 0,5 LA. Si les critères de
performance ne sont pas observés, la méthode devra être
mise en pratique et ses caractéristiques de performance (Q, CVAtyp,
CVLtyp) devront être reétablies durant la nouvelle
validation partielle de la méthode. |
|||
|
4.4. Changements dans l'application de la méthode |
|||||||
|
Changement |
Paramètres à tester |
Pour les méthodes d'essai et les critères d'acceptabilité,
voir les sections appropriées de l'annexe 1. |
|||||
|
4.4.1 Colonne de chromatographie |
Tester la sélectivité de la séparation, de la résolution,
de l'inertie et des valeurs tRR |
Les caractéristiques de performance ne devraient pas être
affectées |
Appliquer des mélanges d'essai appropriés afin d'obtenir
des renseignements sur la performance de la colonne. |
||||
|
4.4.2 Equipement pour traitement échantillons |
Homogénéité de l'échantillon traité;Stabilité
des analytes |
Effectuer les essais décrits en 1.6 et 1.7; ils devraient donner
des résultats conformes aux critères pertinents. |
On procédera à l'essai d'homogénéité
uniquement si le produit est moins bien haché et/ou mélangé
que le produit original. Il faudra tester la stabilité des analytes
si la durée et la température du traitement ont sensiblement
augmenté. |
||||
|
4.4.3 Equipement pour l'extraction |
Comparer les concentrations de résidus d'origine détectés
avec l'ancien et le nouvel équipement dans (5 répliques |
La moyenne des résidus ne devrait pas être très différente
à la concentration p=0,05. |
Essai nécessaire si un nouveau type d'équipement est utilisé |
||||
|
4.4.4 Détection |
Tester la sélectivité de la séparation et la sélectivité
et la sensibilité de la détection |
Les caractéristiques de performance devraient être les mêmes
ou supérieures à celles spécifiées dans la
description de la méthode. |
Tester aussi la détectabilité séparément
avec de nouveaux réactifs de détection. |
||||
|
4.4.5 Analyste |
(5 essais de récupération pour chaque concentration (CEPF,
LA et 2 (3) fois la LA), nouvelle analyse d'un échantillon à
blanc et de deux échantillons positifs (non connus de l'analyste) |
Tous les résultats devraient être compris dans les limites
d'avertissement fixées pour la méthode dans le laboratoire.L'analyse
des échantillons répliques sera faite dans la fourchette
critique. |
Il s'agit d'une prescription minimale. Certains laboratoires utilisent
un protocole plus détaillé qui comprend: (1) production
d'une courbe type dans les critères d'acceptabilité; (2)
au minimum 2 analyses pour chaque matrice contenant des analytes représentatifs
enrichis par l'analyste à un minimum de 3 concentrations dans la
réplique; (3) au minimum une analyse avec des échantillons
enrichis ou avec des résidus d'origine, 3 concentrations dans la
réplique non connus de l'analyste. Tous les résultats doivent
répondre aux critères d'acceptabilité ou être
répétés. |
||||
|
4.4.6 Laboratoire |
Justesse et précision (3 essais de récupération
à chaque concentration (CEPF, LA et 2 (3) fois la LA) par différents
analystes, des jours différents. |
Tous les résultats devraient être compris dans les limites
d'avertissement fixées pour la méthode dans le laboratoire. |
La reproductibilité de la méthode dans les nouvelles conditions
doit être établie, si possible par plus d'un analyste. |
||||
|
Groupe de produits |
Propriétés communes |
Classe de produits[29] |
Espèce représentée |
|
Produits végétaux |
|
|
|
|
I. |
Forte teneur en eau et en chlorophyle |
Légumes feuillus |
Epinard ou laitue |
|
II. |
Forte teneur en eau et peu ou pas de chlorophyle |
Fruits à pépins, |
Pomme, poire pêches, cerises |
|
III. |
Forte teneur en acide |
Agrumes |
Orange, citron |
|
IV. |
Forte teneur en sucre |
|
Raisins, dates |
|
V. |
Riche en huile ou en graisse |
Oléagineux |
Avocat, graines de tournesol, noyers, noix pacane, pistaches |
|
VI.
|
Matières sèches |
Céréales |
Blé, riz ou maïs en grains |
|
|
Produits céréaliers |
Son de blé, farine de blé |
|
|
Produits nécessitant un essai individuel |
|
Par exemple thé, ail, houblon, thé, épices, grosse
canneberbe d'Amérique |
|
|
Produits d'origine animale |
|
||
|
|
|
Viandes |
Viande de bovins, viande de volaille |
|
|
|
Abats comestibles |
Foie, rognons |
|
|
|
Graisses |
Graisse de viande |
|
|
|
Laits |
Lait de vache |
|
|
|
Oeufs |
Oeuf de poule |
Note: La méthode devrait être validée à l'aide de pesticides représentatifs pour chaque groupe de produits. Pour les produits difficiles à analyser, on procédera à des essais individuels.Tableau 6. Exemples de méthodes de détection convenant pour les épreuves de confirmation des substances
|
Méthode de détection |
Critère |
|
CL ou CG et spectrométrie de masse |
Si on contrôle un nombre suffisant d'ions de
diagnostic |
|
CL-DAD ou exploration par UV |
Si le spectre UV est caractéristique |
|
CL - fluorescence |
Associée à d'autres techniques |
|
2-D CCM - (spectrophotométrie) |
Associée à d'autres techniques |
|
CG-DCE, DTI, DPF |
Seulement si associées à une ou plusieurs
techniques de séparation1 |
|
Production de dérivés |
Si ce n'était pas la première méthode de
choix |
|
CL-immunogramme |
Associée à d'autres techniques |
|
CL-UV/VIS (une seule longueur d'onde) |
Associée à d'autres techniques |
1. D'autres systèmes chromatographiques (appliquant des phases stationnaires/mobiles de sélectivité différente) ou d'autres techniques.Glossaire de termes
|
Limite acceptée (LA) |
Valeur de concentration pour un analyte correspondant à
une limite réglementaire ou à une valeur de
référence qui constitue l'objet de l'analyse, par exemple LMR,
LMP; norme commerciale, limite de concentration visée (évaluation
de l'exposition d'origine alimentaire), niveau d'acceptation (environnement),
etc. Pour une substance sans LMR ou pour une substance interdite, il peut ne pas
y avoir de LA (en réalité, elle peut être de zéro ou
elle peut faire défaut) ou il peut s'agir de la concentration
visée au-dessus de laquelle les résidus détectés
devraient être confirmés (limite d'action ou limite
administrative). |
|
Exactitude |
Etroitesse de l'accord entre un résultat
d'expérience et la valeur de référence
acceptée. |
|
Erreur alpha (a) |
Probabilité que la concentration de l'analyte dans
l'échantillon de laboratoire est inférieure à une valeur
particulière (par exemple la LA) lorsque les mesures effectuées
sur une ou plusieurs portions à analyser/d'essai indiquent que la
concentration dépasse cette valeur (faux positif). Les valeurs
acceptées pour cette probabilité sont généralement
de l'ordre de 1 à 5%. |
|
Analyte |
Substance chimique recherchée ou
déterminée dans un échantillon. |
|
Homogénéité de l'analyte (dans
l'échantillon) |
Uniformité de la dispersion de l'analyte dans la matrice. La variabilité dans les résultats d'analyse due à la transformation de l'échantillon est fonction de la dimension de la portion à analyser. La constante d'échantillonnage[30] décrit le rapport entre la dimension de la portion à analyser et la variation prévue dans un échantillon d'analyse bien mélangé: KS = w
(CVSp)[31], où w est la masse
de la portion à analyser et CVSp est le coefficient de
variation de la concentration de l'analyte dans des portions à analyser
répétées de w (g) qui sont prélevées sur
l'échantillon analytique. |
|
PORTION A ANALYSER |
Quantité représentative d'un matériau
prélevée sur l'échantillon à analyser et dont la
taille convient pour la mesure de la concentration de résidu. |
|
ECHANTILLON A ANALYSER |
Matériau préparé pour l'analyse à
partir de l'échantillon de laboratoire, par séparation de la
portion du produit à analyser, puis par mélange, broyage, hachage,
etc. dans le but de prélever des portions à analyser avec une
erreur d'échantillonnage minimale. |
|
APPLICABILITE |
Les analytes, matrices et concentrations pour lesquelles il a
été démontré qu'une méthode d'analyse est
satisfaisante. |
|
Erreur beta (b) |
Probabilité que la concentration effective de l'analyte
dans l'échantillon de laboratoire est supérieure à une
valeur particulière (par exemple la LA) lorsque des mesures prises sur
une ou plusieurs portions à analyser indiquent que la concentration ne
dépasse pas cette valeur (faux négatif). Les valeurs
acceptées pour cette probabilité sont généralement
de l'ordre de 1 à 5%. |
|
Biais |
Différence entre la valeur moyenne mesurée pour
un analyte et une valeur de référence acceptée pour
l'échantillon. Le biais est l'erreur systématique totale par
opposition à l'erreur aléatoire. Il peut y avoir un ou plusieurs
éléments d'erreur systématiques contribuant au biais. Une
différence systématique importante par rapport à la valeur
de référence acceptée se traduit par une valeur de biais
plus élevée. |
|
Groupe de produits |
Groupe de produits destinés à la consommation
humaine ou animale ayant en commun des caractéristiques chimiques
suffisantes qui les rendent similaires aux fins d'analyse par une
méthode. Les caractéristiques peuvent être fondées
sur des constituants importants (par exemple, teneur en eau, graisse, sucre et
acide) ou sur des rapports biologiques, et peuvent être définies
par des règlements. |
|
Méthode de confirmation |
Méthode qui fournit des informations complètes ou complémentaires permettant d'identifier l'analyte avec un degré acceptable de certitude [à la limite acceptée ou niveau qui intéresse l'analyste]. Autant que possible, les méthodes de confirmation fournissent des informations sur les propriétés chimiques de l'analyte, de préférence à l'aide de techniques de spectrométrie. Si une technique particulière ne présente pas une spécificité suffisante, on peut recourir à des procédés supplémentaires, par exemple en combinant de manière appropriée purification, séparation chromatographique et détection sélective. Les titrages biologiques peuvent aussi fournir quelques données de confirmation. Outre l'identité de l'analyte, il faudra aussi
confirmer sa concentration, par exemple par l'analyse d'une deuxième
prise d'essai et/ou d'une nouvelle analyse de la prise d'essai initiale à
l'aide d'une autre méthode appropriée (par exemple colonne ou
détecteur différents). La confirmation qualitative et quantitative
peut aussi être effectuée à l'aide de la même
méthode, le cas échéant. |
|
Limite de décision (CCa) |
Limite à laquelle on peut décider que la concentration de l'analyte présent dans un échantillon dépasse effectivement cette limite, avec une probabilité d'erreur de a (faux positif). Dans le cas d'une substance pour laquelle la limite acceptée est zéro, la CCa est la concentration la plus basse, à laquelle une méthode peut faire la différence avec une probabilité statistique de 1 - a si l'analyte identifié est présent. La CCa équivaut à la limite de détection (LD) selon certaines définitions (en général pour a = 1%). Dans le cas de substance ayant une LA établie, la
CCa est la concentration mesurée, au-dessus
de laquelle on peut décider avec une probabilité statistique de 1
- a que la concentration de la substance à
doser est effectivement supérieure à la LA. |
|
Capacité de détection (CCß) |
C'est la concentration effective la plus faible de l'analyte pouvant être détectée, identifiée et quantifiée dans un échantillon avec une erreur bêta (faux négatif). Dans le cas de substances interdites, la CCß est la concentration la plus faible à laquelle une méthode est capable de déterminer l'analyte dans des échantillons contaminés avec une probabilité statistique de 1 - ß. Dans le cas de substances pour lesquelles une LMR a été fixée, CCß est la concentration à laquelle la méthode est capable de détecter des échantillons qui dépassent cette LMR avec une probabilité statistique de 1 - ß. Quand il est appliqué à la concentration
détectable la plus faible, ce paramètre vise à fournir une
information équivalente à la limite de quantification (LQ), mais
CCß est toujours associée à une probabilité
statistique spécifiée de détection, ce qui explique qu'on
la préfère à la LQ. |
|
Mélange d'essais de détection |
Mélange de substances étalons qui permettent de
vérifier les conditions de séparation chromatographique et de
détection. Le mélange d'essais de détection devrait
contenir des analytes fournissant des informations sur la
sélectivité et les facteurs de réponse pour les
détecteurs, l'inertie (caractérisée par exemple par le
facteur de traîne fT) et la capacité de séparation (par
exemple la résolution Rs) de la colonne, ainsi que sur la
reproductibilité du tRR. Le mélange d'essais de détection
pourrait devoir être spécifique de la colonne et du
détecteur. |
|
Faux résultat négatif |
Voir erreur bêta |
|
Faux résultat positif |
Voir erreur alpha |
|
Résidus d'origine |
Résidus d'un analyte dans une matrice provenant de la
voie par laquelle les résidus à l'état de traces devraient
normalement parvenir, par opposition aux résidus provenant de
l'enrichissement d'échantillons en laboratoire. Appelés aussi
résidus météorisés. |
|
Méthode individuelle |
Méthode apte à déterminer la
présence d'un ou de plusieurs composés spécifiés.
Une méthode individuelle distincte peut être nécessaire, par
exemple pour déterminer certains métabolites inclus dans la
définition des résidus d'un pesticide ou d'un médicament
vétérinaire particulier. |
|
Echantillon de laboratoire |
L'échantillon tel qu'il arrive au laboratoire (non
compris l'emballage). |
|
Limite de détection (LD) |
La plus petite concentration à laquelle l'analyte peut
être identifié. Défini communément comme la plus
petite concentration d'analyte dans la prise d'essai pouvant être
mesurée avec une probabilité établie que l'analyte est
présent à une concentration supérieure à celle de
l'échantillon témoin. L'UICPA et l'ISO ont recommandé le
sigle LD. Voir aussi limite de décision. |
|
Limite de quantification (LQ) |
Concentration la plus faible de l'analyte qui peut être
quantifiée. Définie communément comme la concentration
minimale de l'analyte dans l'échantillon d'essai pouvant être
mesurée avec une précision (répétibilité) et
une exactitude acceptables dans les conditions de l'essai. Voir aussi
capacité de détection. |
|
Concentration étalonnée la plus faible
(CEPF) |
Concentration la plus faible de l'analyte
détectée et mesurée dans l'étalonnage du
système de détection. Elle peut être exprimée comme
une concentration de la solution dans la prise d'essai ou en tant que masse et
ne doit pas comprendre la contribution du témoin. |
|
Matrice |
Matériau ou composant échantillonné
à des fuis d'analyse, à l'exclusion de l'analyte. |
|
Matrice témoin |
Echantillon dans lequel les analytes recherchés ne sont
pas détectables. |
|
Etalonnage ajusté à la matrice |
Etalonnage à l'aide de pesticides de
référence préparés dans un extrait du produit
analysé (ou d'un produit représentatif). Il s'agit de neutraliser
les effets des co-extractifs sur la méthode de détermination. Ces
effets sont souvent imprévisibles, mais l'ajustement à la matrice
peut être inutile lorsque les co-extractifs ont un effet
négligeable. |
|
Méthode |
Série d'opérations depuis la réception
d'un échantillon à analyser jusqu'à la production du
résultat final. |
|
Validation de la méthode |
Procédé visant a vérifier qu'une
méthode est adaptée a l'objectif. |
|
Méthode multi-résidus, MRM |
Méthode convenant pour l'identification et la
quantification d'une gamme d'analytes, habituellement dans un certain nombre de
matrices différentes. |
|
Résultat négatif |
Résultat indiquant que l'analyte n'est pas
présent à ou au-dessus de la concentration étalonnée
la plus basse (voir aussi limite de détection) |
|
Vérification des performances |
Séries de données sur le contrôle de la
qualité produites durant l'analyse des lots d'échantillons pour
valider les analyses en cours. Les données peuvent être
utilisées pour affiner les paramètres de performance de la
méthode. |
|
Résultat positif |
Résultat indiquant que l'analyte est présent
à une concentration à ou au-dessus de la concentration
étalonnée la plus basse. |
|
Précision |
Etroitesse de l'accord entre des résultats d'essais
indépendants obtenus dans des conditions stipulées. |
|
Méthode quantitative |
Méthode pouvant donner des résultats,
exprimés en valeurs numériques dans des unités
appropriées, avec une exactitude et une précision
appropriées à l'objectif. Le degré d'exactitude et de
précision doit être conforme aux critères
spécifiés au tableau 3. |
|
Récupération |
Fraction ou pourcentage d'un analyte
récupéré après extraction et analyse d'une prise
d'essai en blanc à laquelle l'analyte a été ajouté
à une concentration connue (échantillon enrichi ou matériau
de référence). |
|
Blanc de réactifs |
Analyse complète effectuée sans inclure
d'échantillons à des fins de contrôle de
qualité |
|
Matériau de référence |
Matière dont une ou plusieurs concentrations d'analyte
sont suffisamment homogènes et bien établies pour être
utilisées pour l'évaluation d'une méthode de mesure, ou
pour attribuer des valeurs à d'autres matériaux. Dans le contexte
du présent document, le terme "matériau de
référence" ne se réfère pas aux matières
utilisées pour l'étalonnage des appareils. |
|
Méthode de référence |
Méthode d'analyse quantitative dont la fiabilité
a été démontrée par une exactitude, une
spécificité, une précision et une capacité de
détection bien établies. Ces méthodes ont
généralement fait l'objet d'études inter-laboratoires et
s'appuient le plus souvent sur la spectrométrie moléculaire. Le
statut des méthodes de référence est valide uniquement si
la méthode est appliquée dans un régime approprié
d'assurance de qualité. |
|
Procédure de référence |
Procédure dont l'efficacité a été
démontrée. Lorsque cela n'est pas possible, une procédure
de référence pourrait être une procédure en
théorie très efficace et fondamentalement différente de
celle à l'essai. |
|
Répétabilité |
Précision dans des conditions de
répétabilité, c'est-à-dire des conditions dans
lesquelles des résultats d'essais indépendants sont obtenus par la
même méthode sur des portions d'essai identiques dans le même
laboratoire, par le même opérateur utilisant le même
équipement dans un court intervalle de temps (ISO 3534-1) |
|
Analyte représentatif |
Analyte choisi pour représenter un groupe d'analytes
qui ont des chances d'avoir le même comportement durant l'application
d'une méthode d'analyse multi-résidus, si l'on en juge par leurs
propriétés physico-chimiques, par exemple structure,
solubilité dans l'eau, Kow, polarité,
volatilité, stabilité hydrolytique, pKa, etc. |
|
Analyte représenté |
Analyte dont les propriétés physico-chimiques
sont comprises dans la gamme des propriétés des analytes
représentatifs. |
|
Reproductibilité |
Etroitesse de l'accord entre les résultats obtenus par
la même méthode sur des portions d'essai identiques avec des
opérateurs différents utilisant des équipements
différents (dans le cadre de la reproductibilité en laboratoire).
De la même manière, lorsque les essais sont effectués dans
différents laboratoires, on obtient la reproductibilité
inter-laboratoires. |
|
Produit représentatif |
Aliment destiné à la consommation humaine ou
animale utilisé pour représenter un groupe de produits à
des fins de validation d'une méthode. Un produit sera
considéré comme représentatif sur la base de la composition
immédiate de l'échantillon, par exemple teneur en eau,
graisse/huile, acide, sucre et chlorophylle, ou de similitudes biologiques des
tissus, etc. |
|
Robustesse |
Capacité d'une méthode de mesure chimique de
limiter les variations de résultats d'essais lorsqu'elle est soumise
à de faibles variations liées à l'environnement, aux
procédures, aux laboratoires, au personnel, etc. |
|
Préparation de l'échantillon |
Procédé utilisé, si nécessaire,
pour convertir l'échantillon de laboratoire en une prise d'essai, en
enlevant les parties (terre, cailloux, os, etc.) ne servant pas pour
l'analyse. |
|
Transformation de l'échantillon |
Le (ou les) procédé(s) (par exemple
découpage, broyage, mélange) utilisés pour rendre la
portion d'essai suffisamment homogène pour ce qui concerne la
distribution de l'analyte, avant le retrait de la partie à analyser.
L'élément transformateur de la préparation doit être
conçu de manière à éviter des changements induits
dans la concentration de l'analyte. |
|
Méthode de dépistage |
Méthode utilisée pour détecter la
présence d'un analyte ou d'une classe d'analytes à ou au-dessus de
la concentration la plus faible recherchée. Elle devrait être
conçue de manière à éviter les faux résultats
négatifs à un degré de probabilité
spécifié (généralement b = 5%). Il est parfois nécessaire de
confirmer les résultats qualitatifs positifs à l'aide
d'épreuves de confirmation ou de référence. Voir Limite de
décision et capacité de détection. |
|
Sélectivité |
Mesure du degré auquel l'analyte a des chances
d'être séparé des autres composants, soit par
séparation (par exemple chromatographie), soit par réponse
relative du système de détection. |
|
Spécificité |
Mesure dans laquelle une méthode fournit des
réponses à partir du système de détection qui
peuvent être considérées propres à
l'analyte. |
|
Addition de solutions étalons |
Procédé par lequel des quantités connues
de l'analyte sont ajoutées à des parties d'un extrait
d'échantillon contenant l'analyte (sa concentration mesurée au
départ étant X), afin de produire de nouvelles concentrations
nominales (par exemple, 1,5X et 2X). On mesure les réponses de l'analyte
produites par les parties enrichies et l'extrait original, et on
détermine la concentration de l'analyte dans l'extrait original (sans
ajouter d'analyte) à partir de la pente et de l'intersection de la courbe
de réponse. Si la courbe de réponse obtenue n'est pas
linéaire, il faut faire preuve de prudence pour interpréter la
valeur de X. |
|
Facteur de traîne |
Mesure de l'asymétrie du pic de la chromatographie;
à 10% de la hauteur maximale du pic, rapport des segments opposés
de la largeur du pic, lorsqu'il est séparé par une ligne verticale
passant par le pic maximal. |
|
Portion d'essai |
Voir "Portion à analyser" |
|
Echantillon d'essai |
Voir "Echantillon à analyser" |
|
Fidélité |
Etroitesse de l'accord entre la valeur moyenne obtenue
à partir d'une large série de résultats d'essais et la
valeur de référence acceptée. |
|
Incertitude de la mesure |
Paramètre unique (en général un
écart-type ou un intervalle de confiance) exprimant la gamme possible de
valeurs autour du résultat mesuré, dans laquelle on prévoit
que la vraie valeur aura un degré établi de probabilité.
Elle devrait prendre en compte tous les effets reconnus agissant sur le
résultat, y compris la précision globale à long terme (dans
les limites de la reproductibilité en laboratoire) de la méthode
complète; le biais de la méthode; les incertitudes concernant le
sous-échantillonnage et l'étalonnage et toutes autres sources
connues de variations dans les résultats. |
|
Cb |
Voir Appendice IV |
|
Cmax |
Voir Appendice 4 |
|
Cmin |
Voir Appendice 4 |
|
CVAtyp |
Voir Appendice 4 |
|
CVLtyp |
Voir Appendice 4 |
|
CVSp |
Voir Appendice 4 |
|
BPL |
Bonnes pratiques de laboratoire |
|
GSM |
Méthode spécifique de groupe |
|
LMR |
Limite maximale de résidus |
|
MRM |
Méthode multi-résidus |
|
tRR |
Valeur de rétention relative pour un pic |
|
Rs |
Résolution de deux pics chromatographiques |
|
EC |
Ecart-type |
|
Sy/x |
Ecart-type des résidus calculés à partir
de la fonction d'étalonnage linéaire |
|
OMS |
Organisation mondiale de la santé |
![]()
![]()
![]()
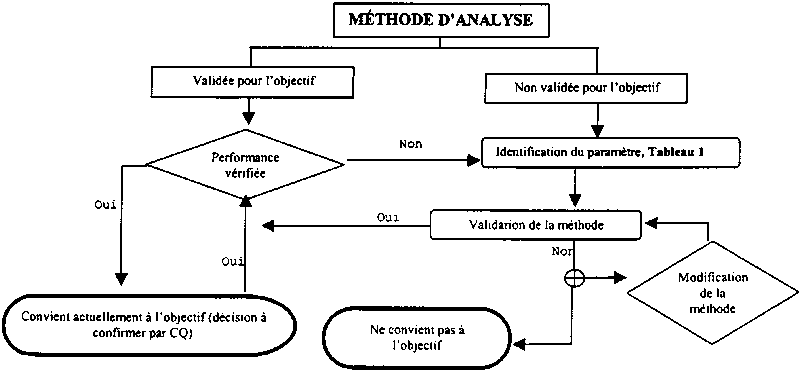{kind=link}
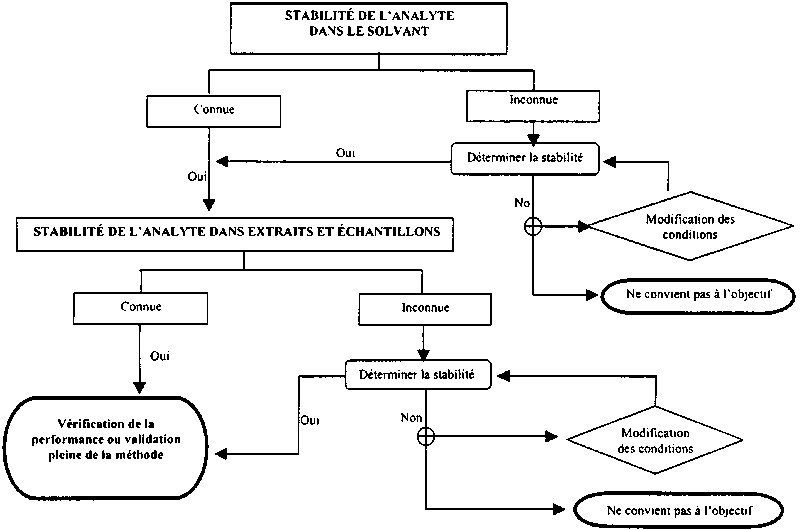{kind=link}