![]()
![]()
![]()
Edward J. Heist
Fisheries and
Illinois Aquaculture Center
Southern Illinois
University at Carbondale
Carbondale, IL,
62901-6511, USA
<[email protected]>
5.1 INTRODUCTION
When members of a fish species are segregated into multiple reproductive stocks, allele frequencies at neutral genetic markers diverge under genetic drift such that the variance in gene frequencies reflects the magnitude of reproductive isolation among these stocks. Thus, gene frequency differences among geographic samples can be used to indirectly estimate patterns of gene flow and hence stock structure of the species. Molecular markers have been used to infer stock structure in fishes for over forty years (Utter, 1991). A brief glossary of genetic terms is included at the end of this section for those readers who may be less familiar with the subject.
Application of molecular markers to the estimation of stock structure in marine elasmobranchs can be challenging for several reasons. Genetic stock structure is less pronounced in marine species, which experience few barriers to migration, than in freshwater species (Ward, Woodwark and Skibinski, 1994). Stock structure is especially weak in highly mobile pelagic fishes (Waples, 1998). Further, sharks exhibit relatively low levels of genetic variation at some molecular markers, perhaps owing to a slowed mutation rate and, or, low long-term effective population sizes (Martin, Naylor and Palumbi, 1992; Smith, 1986). Markers that are not sufficiently variable will not provide the necessary data for a statistically powerful test of stock structure and fish from two geographic regions that are fixed for the same allele may not necessarily be members of the same stock.
The choice of molecular marker depends on the quality and type of tissue available as well as the equipment and expertise. Even a small amount of reproductive migration among stocks is sufficient to prevent genetic divergence at neutral molecular markers. Thus, stocks that are independent from the fisheries perspective may exhibit negligible genetic differentiation (Waples, 1998). Traditional tag and recapture studies performed in concert with molecular genetics studies can provide more information than either approach can individually.
5.2 ESTIMATING STOCK STRUCTURE WITH MOLECULES
The degree to which stocks are reproductively isolated is typically estimated using various forms of Sewell Wright’s FST statistic (Wright, 1931). In the case of a co-dominant locus that exhibits only two alleles FST is equal to
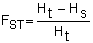 | (5.1) |
where Ht is the expected heterozygosity in the population based on the mean allele frequency across populations and expectations of Hardy-Weinberg equilibrium (i.e. Ht = 2pq where p = the frequency of one allele and q = 1-p) and HS is the mean heterozygosity within populations. Thus, the greater the variance in allele frequencies among populations the greater the deficit of heterozygosity within each population. FST can be determined directly from the variance in allele frequencies as:
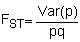 | (5.2) |
where Var(p) is the variance in the frequency of an allele among subpopulations. Expected values of FST range from zero when each sample possesses identical gene frequencies and hence there is a single genetic stock, to unity when isolated stocks are fixed for alternate alleles.
Either of these measures is sensitive to sampling error and in the absence of distinct stocks will result in positive FST values, the magnitudes of which are inversely proportional to sample size. Waples (1998) observed that in highly migratory species, such as many sharks, the magnitudes of FST estimates resulting from sampling error alone may be larger than the parametric FST values among stocks. Various unbiased estimators of Wright’s FST include Weir and Cockerham’s θ estimator, which includes corrections for several types of sampling error and sometimes produces negative FST estimates when the true value of FST is small or zero (Weir and Cockerham, 1984). Many recent studies employ analysis of molecular variation (AMOVA) (Excoffier, Smouse and Quattro, 1992), which provides an unbiased estimator of FST known as ΦST and also permits partitioning of genetic variation to multiple hierarchical levels. These estimators are computationally demanding but are incorporated into a variety of freely available software packages. Statistical tests of the hypothesis that ΦST = 0 (and hence samples are drawn from a single genetic stock) are calculated using algorithms that either model or resample the data and determine the significance level of ΦST as the likelihood that a larger ΦST value could be produced via a random allocation of the genotypes or alleles (Rousset, 2001).
Several software packages are freely available for analyzing molecular genetic data including Arlequin (http://lgb.unige.ch/arlequin/) (Schneider, Roessli and Excoffier, 2000), Genepop (http://wbiomed.curtin.edu.au/genepop/) (Raymond and Rousset, 1995), and GDA (http://lewis.eeb.uconn.edu/lewishome/software.html) (Lewis and Zaykin, 2001). The capabilities of these and several other programs were recently reviewed by Labate (2000). Arlequin can be downloaded in Microsoft Windows, Macintosh or Linux format and can handle haploid (e.g. mtDNA) as well as diploid (allozyme and microsatellite) data. Genepop can be downloaded to run in a windows environment or can be run directly from the web page. GDA is only available in windows format and determines significance of θ by bootstrapping across loci, which is only applicable to studies that employ a large number of loci. Under the assumptions of the island model of migration (Wright, 1931), which assumes a large number of discrete populations with equal amounts of migration among each population, FST can be related to migration as:
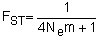 | (5.3) |
where Nem is the product of the effective population size and the migration rate. Nem can be thought of as the effective number of migrants, that is the number of reproductive animals exchanged among populations. It may seem counterintuitive that the magnitude of FST would be related to the number of migrants and not migration rate. However, the degree to which allele frequencies among isolated populations diverge due to genetic drift is inversely proportional to the effective population size. Thus, populations with a large Ne require a smaller migration rate to produce the same magnitude of genetic variance among populations (FST ). The relationship implies several simplifying assumptions that are unrealistic for shark populations (e.g. equal migration among each of the many populations). However, deviations from these assumptions have only minor effects on the relationship between FST and Nem. For example, the more realistic case of increased migration among geographically proximate locations and a small number of populations produces slightly lower FST values for the same rate of migration (Mills and Allendorf 1996).
Mitochondrial (mt) DNA is potentially a more powerful marker than nuclear DNA. Because mtDNA is maternally inherited as a haploid molecule it has approximately ¼ the effective population size of a nuclear marker (Birky, Marayama and Fuerst, 1983). The relationship between FST and migration is
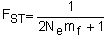 | (5.4) |
where Nemf refers to the effective migration rate of females only. In species with equal rates of male and female migration the magnitude of FST will be greater for mitochondrial markers than nuclear markers. Further, because of the smaller effective population size mtDNA reaches equilibrium levels of FST more quickly and thus a recently established pattern of stock structure will be more accurately represented by mitochondrial data than by nuclear DNA data. In species that exhibit female reproductive philopatry and outcrossing with males from widespread localities, such as several species of marine mammals and sea turtles, mtDNA exhibits stronger differentiation than nuclear markers (Karl and Bowen, 1992; Palumbi and Baker, 1994; and Gladden et al., 1999). However, the differences in the rates of genetic drift, mutation and intraspecific variation among mitochondrial and nuclear markers are sufficient to produce vast differences in estimates of FST between the marker types without any differences in male and female-mediated gene flow (Buonaccorsi, McDowell and Graves, 2001). Thus, larger FST values for mitochondrial markers relative to nuclear markers do not necessarily indicate female philopatry against a backdrop of male roaming.
5.3 MOLECULAR MARKERS
5.3.1 Marker type
Several types of molecular markers have been applied to the estimation of stock structure in sharks and many other types used in other marine fishes have yet to be employed in elasmobranchs. The choice of marker depends on the experience of the researcher and the types of equipment and the types and quality of tissue that are available. It is unfeasible to provide specific protocols here, but several excellent published volumes containing protocols for these and other techniques include Hillis, Moritz and Mable (1996), Ferraris and Palumbi (1996) and Hoelzel (1998).
5.3.2 Allozymes
Allozymes were the first molecular markers to gain widespread use for distinguishing stocks of fishes (Utter, 1991). Allozymes are distinct allelic forms of enzymes that are separated by charge and in some cases three-dimensional shape on a separatory medium, typically starch gels, polyacrylamide gels or cellulose acetate plates and visualized with histochemical stains that indicate the migration of molecules with specific enzyme activities (Murphy et al., 1996; May, 2003). Allozymes degrade rapidly after death, especially at high temperatures, and the use of allozymes as molecular markers requires fresh or frozen tissue (maintained att -20°C or preferably colder). Because tissue types vary in enzyme expression, it is often useful to collect multiple tissue types (e.g. white muscle, heart, liver, brain) to score a large number of loci. Thus, allozyme electrophoresis is not the best technique where lethal sampling and immediate freezing (e.g. with dry ice or liquid nitrogen) of tissue samples are not possible.
Resolution of allozyme banding patterns requires considerable interpretation (Buth, 1990). Homozygotes for different alleles produce single bands with varying motilities while heterozygotes take on an appearance that is determined by the subunit structure of the active enzyme. Monomeric enzymes produce two-banded heterozygotes while dimeric and tetrameric enzymes (those possessing two and four peptides per active enzyme) exhibit three-and five-banded heterozygotes. Many enzymatic reactions are catalyzed by products of multiple loci heteropolymers, which can further complicate the banding patterns. Resolution of allozyme patterns as discrete bands rather than smears requires the screening of multiple running buffer conditions to identify the optimal conditions for each locus.
Several studies of allozymes have detected low levels of variation in sharks. In the first published study of allozymes in sharks Smith (1986) reported relatively low variation in seven species. Low levels of allozyme variation and geographic heterogeneity in carcharhinid sharks were observed by Lavery and Shaklee (1989) and by Heist, Graves and Musick (1995). Relatively high levels of heterozygosity and heterogeneity were found in Pacific angel sharks (Squatina californica) (Gaida, 1997) and gummy sharks (Mustelus antarcticus) (Gardner and Ward, 2002).
Resolution of allozyme loci can be more of an art than a science and variation in the methodology and experience among labs result in differences in the amount of variation that can be resolved on allozyme gels. Gardner and Ward (1998) found that on average 25.5% of allozyme loci in gummy shark were polymorphic with a mean heterozygosity of 0.099. For the same species over a somewhat smaller geographic range MacDonald (1988) detected variation in only one of 32 presumed loci (3%) with a mean heterozygosity of 0.006 in the same species. Certainly some of this discrepancy must be due to the increased resolution of the study by Gardner and Ward.
The relative simplicity of the materials needed to perform the allozyme technique (i.e. many rigs are “homemade”) make allozymes an attractive tool for labs with little research funding. However, as PCR-based techniques are becoming more affordable, the low variation and high tissue quality demands of allozymes make techniques that score variation at the DNA-level more attractive. Plans for allozyme equipment can be found in Aebersold et al.(1987) and Murphy et al.(1996).
5.3.3 Mitochondrial DNA
Mitochondrial DNA of elasmobranchs and other fishes is a single closed loop of double stranded DNA approximately 16500 base pairs (bp) in length and presumably inherited only from the maternal parent (Billington, 2003). The haploid, uniparental inheritance of mtDNA results in a fourfold reduction in the effective population size and therefore an accelerated rate of genetic drift, which in turn increases the rate and magnitude of genetic differentiation among isolated fishery stocks (Birky, Marayama and Fuerst, 1983). Data derived from sequencing or restriction fragment length polymorphism (RFLP) analysis of mtDNA permit estimation of the relative divergence time of any two mtDNA haplotypes and can be used to provide evidence of deep historic divisions or cryptic species (Figure 5.1).
If relatively large quantities (several grams) of fresh or ultrafrozen tissue and an ultracentrifuge are available, mtDNA can be isolated in its pure circular form and be subjected to restriction enzymes that cleave the circular DNA at specific four-to six-base motifs. The resultant population of restriction fragments can be resolved on agarose or polyacrylamide gels and seen using radiolabeling or UV illumination of ethidium bromide stained bands (Figure 5.1). This technique was performed by Heist, Graves and Musick (1995) Heist, Musick and Graves (1996a, 1996b) on sandbar (Carcharhinus plumbeus), shortfin mako (Isurus oxyrinchus) and Atlantic sharpnose (Rhizoprionodon terraenovae) sharks. In the sharpnose shark study, whole molecule mtDNA prepared from tiger shark was used to probe southern blots of Atlantic sharpnose shark hearts that did not provide sufficient whole-molecule mtDNA.
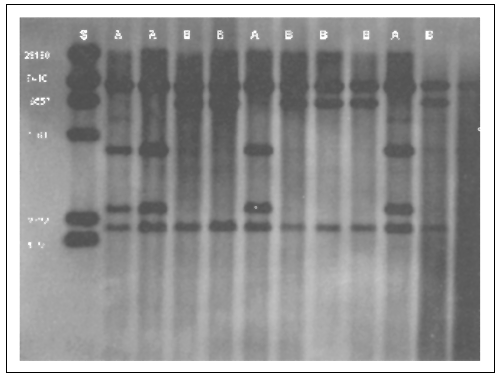
FIGURE 5.1
Mitochondrial DNA variation in shortfin mako (Isurus
oxyrinchus). Lane “S”is a size standard lane. Numbers at left refer to the size
(in base pairs) of each size standard. Whole molecule mtDNA digested with the
restriction enzyme Bst E II produces two haplotypes (A and B). Haplotype
“A”differs from “B”in that a fragment of approximately 7000 base pairs in “B”is
digested into two smaller fragments of approximately 4400 and 2600 in “A”.
With the advent of PCR more studies are employing restriction digestion or sequencing of discrete regions of mtDNA. Perhaps the most useful region for analyzing stock structure in elasmobranchs is the D-loop or control region, which contains the largest stretches of noncoding DNA in the elasmobranch mtDNA genome and in many fishes studied it exhibits the highest nucleotide substitution rate, presumably due to the lack of purifying selection. In my lab we routinely use a primer designed by Martin and Palumbi (1993) located in the cytochrome-b protein coding region (CB6H 5’CTC CAG TCT TCG RCT TAC AAG where “R”represents equal quantities of A and G) and a mammalian primer designed in the highly-conserved 12S ribosomal gene (282 5’AAG GCT AGG ACC AAA CCT) (J.C. Patton, LGL Alaska Research Associates, Anchorage, USA, unpublished data) to amplify the entire D-loop region in a variety of sharks. The resultant PCR product can then be analysed using restriction enzymes or direct sequencing. The widespread availability of inexpensive thermal cyclers and gel rigs make PCR-RFLP a viable method of analysis for labs with a limited research budget.
The genetic diversity present in mtDNA can be represented as haplotype diversity which is estimated as
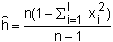 | (5.5) |
where h is the haplotype diversity, n is the number of individuals scored, xi is the frequency of each allele, and l is the number of unique haplotypes detected (Nei and Tajima 1981). This equation is essentially the same as that for estimating heterozygosity at a diploid locus and can be thought of as the likelihood that two randomly sampled haplotypes differ. Because haplotype diversity is affected by the number of bases surveyed (i.e. amount of sequence data or number of restriction enzymes employed) a more universal gauge of variation is nucleotide sequence diversity (δ) which can be estimated as
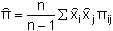 | (5.6) |
Where 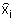 and
and 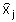 are the frequencies of haplotypes i and j and
are the frequencies of haplotypes i and j and
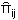 is the genetic distance between each pair of
haplotypes (Nei and Tajima 1981). AMOVA (Excoffier, Smouse and Quattro, 1992)
can then be used to estimate ΦST by partitioning the genetic diversity into among and between
sample components. The REAP software package (McElroy et al., 1991), which is
available at http://bioweb.wku.edu/faculty/mcelroy/, can be used to estimate ŏ
and to construct a distance matrix between haplotypes for AMOVA.
is the genetic distance between each pair of
haplotypes (Nei and Tajima 1981). AMOVA (Excoffier, Smouse and Quattro, 1992)
can then be used to estimate ΦST by partitioning the genetic diversity into among and between
sample components. The REAP software package (McElroy et al., 1991), which is
available at http://bioweb.wku.edu/faculty/mcelroy/, can be used to estimate ŏ
and to construct a distance matrix between haplotypes for AMOVA.
Sharks possess relatively low levels of intraspecific mtDNA heterogeneity, presumably due to the low rate of mtDNA evolution relative to that of other vertebrates (Martin, Naylor and Palumbi, 1992). Levels of nucleotide sequence diversity based on whole-molecule RFLP in sharks range from 0.036% in sandbar shark (Heist, Graves and Musick 1995) to 0.347% in shortfin mako (Heist, Musick and Graves, 1996a). To detect a sufficient amount of variation one must either perform the whole-molecule technique with a large number (e.g. eight or more) restriction enzymes or perform direct sequencing. In our lab we are sequencing the entire mtDNA D-loop in blacktip sharks (C. limbatus) to produce a haplotype diversity of 0.71 (Keeney et al., 2003).
5.3.4 Microsatellites
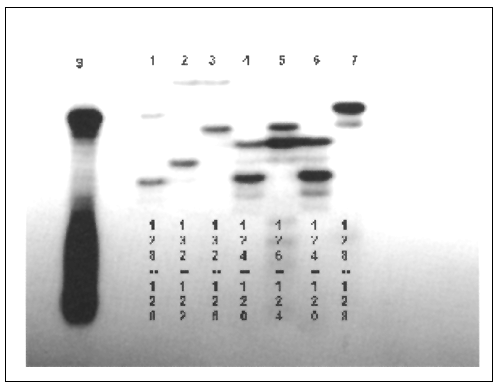
DNA microsatellites are among the most recent types of markers developed for estimating stock structure. These are highly repetitive segments of nuclear DNA that are amplified via PCR and typically resolved on polyacrylamide gels (O’Connell and Wright, 1997). Microsatellite alleles differ in size based upon differences in the number of repeat units present. Alleles differ in size by multiples of the core repeat motif (typically two to four bases) and thus high resolution is required to score microsatellites. Typically PCR products are end-labeled with radionuclides (e.g. 32P or 33P) and resolved via autoradiography (Figure 5.2) or fluorescently tagged and resolved on automated DNA sequencers. Both of these techniques may be beyond the capabilities of labs with limited budgets and, or without access to radionuclides.
FIGURE 5.2
Microsatellite DNA variation in nurse shark (Ginglymostoma
cirratum). Lane “S”is a 128 base pair size standard. Individuals 1 through 6
are heterozygous (genotypes shown below bands). Individual 7 is homozygous for
allele 128.
The major hurdle to scoring microsatellites in any species is the development of PCR primers that amplify polymorphic loci. To date polymorphic microsatellite loci have been developed in sandbar shark (Heist and Gold, 1999), white shark (Carcharodon carcharias) (Pardini et al., 2000), lemon shark (Negaprion brevirostris) (Feldheim, Gruber and Ashley, 2001a; 2001b), shortfin mako (Schrey and Heist, 2002) and nurse shark (Ginglymostoma cirratum) (Heist et al., 2003). Primers developed in one species often work on congeners and sometimes members of related genera but either fail to amplify or amplify only monomorphic products in other families or in more distantly-related taxa. Of sixteen polymorphic microsatellite loci developed from the blacktip shark between five and eleven loci were polymorphic in each of ten other species of Carcharhinus and several loci were polymorphic in tiger shark, lemon shark, blue shark (Prionace glauca), Atlantic sharpnose shark and two species of hammerhead sharks (Sphyrna spp.) (Keeney and Heist, 2003). Primers developed in shortfin mako amplified polymorphic microsatellites in salmon (Lamna ditropis), porbeagle (L. nasus) and white sharks (Schrey and Heist,2002).
Microsatellite data are analysed much like allozyme data although the high heterozygosity and large number of alleles (e.g. 20 or more) can cause a deflation of FST (Hedrick, 1999). Microsatellites evolve via mutational increases and decreases in the number of times the core motif is repeated in each allele. Thus, microsatellites exhibit a finite number of alleles and alleles are often shared even among completely isolated gene pools (e.g. among species). The maximum value FST can be expected to achieve is equal to homozygosity, which for loci with 20 or more alleles may be less than 0.05. Thus, the maximum value that can be achieved for FST is comparable to the expected error associated with measurements involving small sample sizes (Waples, 1998). An obvious way to alleviate some of this problem is to employ loci with moderate numbers of alleles and moderate heterozygosities and to obtain sufficiently large sample sizes to reduce the error in estimates of FST .
A common problem that attends the high genetic diversity of microsatellites and the sampling power of modern estimators is the detection of small but statistically significant FST values. Low but significant FST values can arise through a small amount of gene flow (e.g. 1–10 individuals per generation) between stocks that are essentially discrete in terms of recruitment, or they can be an artifact of sampling (e.g. inclusion of close relatives in a sample) and scoring (e.g. null alleles) and thus constitute a Type I error. Dizon, Taylor and O’Corry-Crowe (1995) warned that the consequences of failing to reject the null hypothesis of FST =0 when it is false (Type II error) may be more deleterious to the management of a species than falsely concluding that multiple stocks are present and recommended that power analyses be used to adjust the rejection (α) level upward to a level that balanced the effects of both types of statistical error. Feldheim, Gruber and Ashley (2001b) concluded that a statistically significant (p< 0.05) θ value of 0.016 based on highly polymorphic (Heterozygosity = 0.69 to 0.90) microsatellite loci was too low to consider lemon sharks from the Florida, the Bahamas and Brazil as distinct stocks. Tagging data (Kohler, Casey and Turner, 1998) indicate that lemon sharks move between the Bahamas and Florida, but no lemon sharks tagged in either Florida or the Bahamas moved to the Caribbean or beyond. Thus, it seems unlikely that lemon sharks from Florida and Brazil do not comprise distinct fishery stocks. While gene flow has apparently been high enough to prevent evolutionary divergence among lemon sharks in the western Atlantic, statistically significant differences in allele frequency, regardless of their magnitude, indicate that samples are drawn from different populations (Knutsen et al., 2003).
5.3.5 Other markers
Several other types of molecular markers are used to assess stock structure in fishes but have yet to be applied to elasmobranchs. Random Amplified Polymorphic DNA (RAPD) employs one or more short primers (typically about ten bases) to amplify a population of fragments that are resolved on agarose or polyacrylamide gels (Hadrys, Balcik and Schierwater, 1992). The degree to which bands are shared among individuals can be used to assess the relatedness of individuals within and among populations. While this method is attractive because it does not require taxon-specific primers as mtDNA RFLP and microsatellites do, there are several serious shortcomings that have prevented this technique from gaining widespread acceptance as a tool for analysis of stock structure. PCR is a finicky process that often produces inconsistent results, especially with short primers and low annealing temperatures. Whether a faint band is present or absent may depend on the quality of the tissue used to prepare the DNA or the dynamics of the specific PCR reaction that produced the profile. If tissue quality varies among sample locations, there can be a systematic bias in the data leading to an erroneous conclusion of stock structure.
Another technique, Amplified Fragment Length Polymorphism (AFLP) analysis, is performed by attaching oligonucleotide adapters to nuclear DNA restriction fragments and amplifying with longer PCR primers that anneal mostly to the adapters, but also the first of three bases of the genomic DNA (Vos et al., 1995). While this approach is far more work than RAPD, the data are more repeatable because of the use of longer PCR primers and higher annealing temperatures. Both RAPD and AFLP produce dominant data (i.e. there is generally no way to distinguish between bands that are present in heterozygous or homozygous dosages) and as a result statistical treatment of the data are not as powerful as those for co-dominant data (e.g. allozymes and microsatellites).
5.3.6 Tissue collection
The kinds of tissue samples available and the method of preservation determine what kinds of molecular markers can be used. PCR-based methods are most forgiving and can even be performed on dried fins (Shivji et al., 2002). For PCR-based analyses we routinely collect fin clips using a scalpel to excise approximately 0.5 cm2 from the trailing edge of the first dorsal fin. The thin trailing edge of the fin produces far better yields of DNA than do muscle tissue or thick skin from other parts of the body. Fin clips can be stored in either 95% ethanol or 20% dimethyl sulfoxide saturated with NaCl. Tissues are stable in either medium at room temperature for several months, however long term storage of ethanolpreserved tissues is best done at 4°C or colder. Tissues for whole molecule RFLP need to be kept fresh or frozen once and not subjected to freeze-thaw cycles as each freezing cycle produces ice crystals that linearize the mitochondria making mtDNA purification difficult. Tissues for allozymes are most demanding in that enzymes degrade rapidly after death. Tissues need to be frozen (preferable in dry ice or liquid nitrogen) and maintained as cold as possible until homogenized for electrophoresis.
Many sharks undergo seasonal and reproductive migrations and may segregate by sex and life stage. Thus, a careful choice of where, when and from which animals to collect tissue can influence the outcome of a study. For example, in the study of blacktip sharks described by Keeney et al. (2003) all tissues were collected from neonate sharks near or within continental nursery areas. Thus, any signal resulting from reproductive philopatry could be filtered from the noise of adult movement. Such studies can be biased because a sample from a single nursery may contain siblings, which would tend to inflate estimates of gene frequency differences among samples. However, because sharks such as the blacktip have low fecundities and do not reproduce every year, the number of potential sibling pairs is low and comparisons across sequential years can be used to determine whether a sampling of siblings is influencing estimates of FST .
5.4 SELECTED CASE STUDIES
5.4.1 Gummy shark
The gummy shark is a small coastal species continuously distributed around the southern two-thirds of Australia. Gardner and Ward (1998) found statistically significant differences in allozyme allele and mtDNA haplotype frequencies in gummy sharks collected from the southern and southeastern coasts of Australia including Tasmania. Measures of GST (an analog of FST ) were significantly greater than the values expected due to sampling error for three of seven polymorphic loci and for RFLP haplotypes of whole-molecule mtDNA. Both molecular markers indicated that gummy sharks from the southern coast of Australia, ranging from Bunbury to Eden and including Tasmania, comprised a single stock while gummy sharks from the east coast of Australia from Eden north comprised one or more additional stocks. Vertebral counts did not differ throughout southern Australia. However, there appeared to be a gradual increase in the number of precaudal vertebrae corresponding to decreasing latitude on the east coast. Thus, despite the continuous distribution and great potential for movement of M. anarcticus, there exist multiple fishery stocks in Australian waters. Subsequently, Gardner and Ward (2002) reported data from additional Mustelus species including M. lenticulatus from New Zealand and two putative but undescribed species from Australia. Allozyme, mtDNA, and vertebral count data all confirmed the presence of four species of Mustelus in the waters of Australia and New Zealand.
5.4.2 Blacktip shark
The blacktip shark is a migratory species that is the most important component of the US longline shark fishery operating in the southeastern United States in the Atlantic Ocean and Gulf of Mexico. Neonate blacktip sharks from the west coast of Florida migrate south in the fall, presumably to southern Florida, and have been shown to return to specific nursery areas in subsequent years (Keeney et al., 2003). Whether adult females return to their natal nurseries for parturition is unknown. A study of mtDNA sequences and microsatellites in young-of-the-year blacktip sharks collected from four nursery areas: west coast of Florida, South Carolina, Texas and Mexican Yucatan revealed significant heterogeneity in mtDNA (FST = 0.111, p<0.001) but not microsatellite loci (FST < 0.001, P=0.316) (Hueter et al., 2005). Neither marker revealed significant differences among three Florida nurseries separated by less than 250 km. Thus, blacktip sharks comprise multiple fishery stocks in US and Mexican waters and while females may tend to return to natal nurseries the fidelity to do so is not high enough to result in significant structuring among proximal nurseries.
5.4.3 White shark
The white shark is a wide-ranging globally distributed species with populations clustered around localities with abundant marine mammals. Pardini et al. (2001) compared mtDNA and nuclear (microsatellite) markers in white sharks from South Africa, Australia and New Zealand. The mtDNA data indicated two divergent clusters of haplotypes that were nearly clustered into two highly divergent clades. One clade (type A) was found in 48 of 49 individuals surveyed in Australia and New Zealand while the other clade was found in 39 individuals from South Africa and in one of the 49 individuals surveyed in the Australia-New Zealand sample. FST analogs (θ) based on five microsatellite loci were all non-significant. Based on the discrepancy in estimates of stock structure between nuclear and mitochondrial data Pardini et al. (2001) concluded that female white sharks are much more philopatric than males.
5.4.4 Shortfin mako
The shortfin mako is a highly migratory cosmopolitan species found throughout the Atlantic, Pacific and Indian Oceans. Heist, Musick and Graves (1996a) examined whole molecule mtDNA RFLP data in 120 shortfin makos from the North Atlantic (US and Canada), South Atlantic (Brazil), North Pacific (California) and South Pacific (Australia) and found small but significant differences in haplotype frequencies between the North Atlantic and all other samples. Subsequently, Schrey and Heist (2003) examined microsatellites in 433 mako sharks including the individuals described in Heist, Musick and Graves (1996a). They also reanalysed the data from Heist, Musick and Graves (1996a) using a more powerful statistical approach. Among-ocean-basin FST estimates from the mitochondrial data were significant and two orders of magnitude larger than the estimates of FST based on microsatellites. A power analysis indicated that if the amount of heterogeneity present in the mtDNA data accurately represented the magnitude of gene flow of both sexes a statistically significant FST would have been detected using microsatellites, assuming that the stock structure was stable long enough for nuclear markers to reach equilibrium. The discrepancy in the levels of resolution in mtDNA and microsatellites is likely due to sex-biased dispersal, but they could also be influenced by differences in the resolving powers of the two markers. The shortfin mako results differed from those of white sharks (Pardini et al., 2002) in that no strong phylogeographic signal is present in the mtDNA data, only minor frequency differences among locations. Shortfin mako does not comprise a single worldwide population, but there has been a sufficient amount of historical migration among ocean basins to make detection of stock structure using molecular markers (and especially nuclear DNA markers) challenging.
5.5 CONCLUSIONS
Using molecular markers to estimate stock structure in sharks can be challenging owing to the great potential for migration among shark stocks, the difficulty in detecting genuine but small differences in gene frequencies in the presence of recent or episodic migration among stocks and inappropriate (too low or too high) levels of variation provided by some molecular markers. Nevertheless several studies have ably demonstrated stocks in sharks and even in highly migratory species across seemingly continuous distributions. Comparisons between markers with different modes of inheritance (e.g. nuclear vs. mitochondrial) may indicate differences in male-versus female-mediated gene flow. Because many sharks are viviparous k-strategists that produce well-formed young at a time and place conducive to survival, stocks that overlap during part of the year may segregate into discrete stocks for mating and, or parturition. Thus, a careful selection of where and when tissues are collected (e.g. from neonates in nursery areas) coupled with a wise choice of a molecular marker can provide valuable information about the stock structure of sharks that cannot be obtained from other methods.
Molecular detection of stock structure is a complementary technique to tagging and morphology based studies of stock structure. While tagging reveals gross movements of individuals, genetics measures the flow of genes over many generations and can be used to study fidelity to nursery or breeding grounds in animals whose distributions may sometimes overlap with those of other stocks. Morphological and life history differences may be due to different environmental influences and hence may, or may not, be reflected in gene frequency differences at neutral loci.
5.6 GLOSSARY OF GENETIC TERMS USED
Allele -Alternate forms of a gene at a particular locus. Each diploid organism may possess either one (homozygote) or two (heterozygote) alleles at a locus; however, there may be more than two alleles in a population.
Co-dominant markers -Markers that exhibit both alleles in a heterozygous state. Co-dominant markers are more powerful than dominant markers in which a heterozygous individual is indistinguishable from an individual homozygous for the dominant allele.
Fixed allelic differences -The absence of shared alleles between two populations.
FST -An index of the magnitude of allele frequency difference among populations. At a locus with two alleles the maximum value of FST is unity and occurs when each population bears only a single allele not found in any other population. If allele frequencies are identical across populations, FST= 0.
Genetic drift -Random change in gene frequencies due to random stochastic sampling of alleles from generation to generation.
Heterozygosity -The fraction of individuals that exhibit two different alleles at a locus or alternately the fraction of loci over which an individual exhibits two different alleles.
Heterozygous -Possessing two different alleles at a locus.
Homozygous -Possessing two identical alleles at a locus.
Locus -A particular location on a chromosome where a gene or other DNA sequence resides. Diploid organisms possess two copies of each locus that may exhibit either the same (homozygote) or different (heterozygote) alleles.
Mitochondrial DNA (mtDNA) -DNA found in the mitochondria in cells. In animals, including sharks, mtDNA is a double stranded molecule approximately 16500 base pairs in length. Mitochondrial DNA is inherited strictly from the female parent and thus is a haploid (one copy per cell) marker.
Molecular marker -A polymorphic heritable trait that can be scored for variation within or between species.
Neutral genetic markers -Polymorphic genetic traits that are presumed not to be influenced by natural selection and thus are sensitive only to mutation, migration and genetic drift. Most models that relate gene frequency differences with stock structure assume that the markers examined are selectively neutral.
Nuclear DNA -The vast majority of DNA in animal cells is found in the nucleus. Nuclear DNA is equally inherited from both parents and thus is a diploid (two copies per cell) marker.
Polymerase Chain Reaction (PCR) -A technique for producing millions of copies of a chosen segment of DNA by repeatedly annealing sequence-specific primers on either side of the region of interest and performing (typically) thirty or more cycles of DNA synthesis. This procedure allows the characterization of a particular segment of nuclear or mitochondrial DNA using only minute amounts of tissue.
5.7 LITERATURE CITED
Aebersold, P.B., Winans, G.A., Teel, D.J., Milner, G.B. & Utter, F. 1987. Manual for starch gel electrophoresis: a method for the detection of genetic variation, NOAA, Washington, D.C., 19 pp.
Billington, N. 2003. Mitochondrial DNA. In E.M. Hallerman (ed.). Population genetics: principles and applications for Fisheries Scientists, pp. 59–100. American Fisheries Society, Bethesda, MD. 457 pp.
Birky, C.W.J., Marayama, T. & Fuerst, P. 1983. An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics, 103: 513–527.
Buonaccorsi, V.P., McDowell, J.R. & Graves, J.E. 2001. Reconciling patterns of inter-ocean molecular variance from four classes of molecular markers in blue marlin (Makaira nigricans). Molecular Ecology, 10: 1179–1196.
Buth, D.G. 1990. Genetic Principles and the interpretation of elecrophoretic data. In D.H. Whitmore (ed.). Electrophoretic and isoelectric focusing techniques in fisheries management, pp. 1–21. CRC Press, Boca Raton, FL. 360 pp.
Dizon, A.E., Taylor, B.L. & O’Corry-Crowe, G.M. 1995. Why statistical power is necessary to link analyses of molecular variation to decisions about population structure. In J.L. Neilson & D.A. Powers (eds). Evolution and the aquatic ecosystem. American Fisheries Society, Bethesda, MD.
Excoffier, L., Smouse, P.E. & Quattro, J.M. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics, 131: 479–491.
Feldheim, K.A., Gruber, S.H. & Ashley, M.V. 2001a. Multiple paternity of a lemon shark litter (Chondrichthyes : Carcharhinidae). Copeia, pp. 781–786.
Feldheim, K.A., Gruber, S.H. & Ashley, M.V. 2001b. Population genetic structure of the lemon shark (Negaprion brevirostris) in the western Atlantic: DNA microsatellite variation. Molecular Ecology, 10: 295–303.
Ferraris, J.D. & Palumbi, S.R. 1996. Molecular zoology: advances, stratgies and protocols. John Wiley and Sons, Inc., New York, USA. 580 pp.
Gaida, I.H. 1997. Population structure of the Pacific angel shark, Squatina californica (Squatiniformes : Squatinidae), around the California Channel Islands. Copeia, pp. 738–744.
Gardner, M.G. & Ward, R.D. 1998. Population structure of the Australian gummy shark (Mustelus antarcticus Gunther) inferred from allozymes, mitochondrial DNA and vertebrae counts. Marine and Freshwater Research, 49: 733–745.
Gardner, M.G. & Ward, R.D. 2002. Taxonomic affinities within Australian and New Zealand Mustelus sharks (Chondrichthyes : Triakidae) inferred from allozymes, mitochondrial DNA and precaudal vertebrae counts. Copeia, pp. 356–363.
Gladden, J.G.B., Ferguson, M.M., Friesen, M.K. & Clayton, J.W. 1999. Population structure of North American beluga whales (Delphinapterus leucas) based on nuclear DNA microsatellite variation and contrasted with the population structure revealed by mitochondrial DNA variation. Molecular Ecology, 8: 347–363.
Hadrys, H.M., Balcik, M. & Schierwater, B. 1992. Application of random amplified polymorphic DNA (RAPD) in molecular Ecology. Molecular Ecology, 1: 55–63.
Hedrick, P.W. 1999. Highly variable loci and their interpretation in evolution and conservation. Evolution, 53: 313–318.
Heist, E.J. & Gold, J.R. 1999. Microsatellite DNA variation in sandbar sharks (Carcharhinus plumbeus) from the Gulf of Mexico and mid-Atlantic Bight. Copeia, 5: 182–186.
Heist, E.J., Graves, J.E. & Musick, J.A. 1995. Population genetics of the sandbar shark (Carcharhinus plumbeus) in the Gulf of Mexico and Mid-Atlantic Bight. Copeia, 18: 555–562.
Heist, E.J., Musick, J.A. & Graves, J.E. 1996a. Genetic population structure of the shortfin mako (Isurus oxyrinchus) inferred from restriction fragment length polymorphism analysis of mitochondrial DNA. Canadian Journal of Fisheries & Aquatic Sciences, 53:583–588.
Heist, E.J., Musick, J.A. & Graves, J.E. 1996b. Mitochondrial DNA diversity and divergence among sharpnose sharks, Rhizoprionodon terraenovae, from the Gulf of Mexico and Mid-Atlantic Bight. Fishery Bulletin, 94: 664–668.
Heist, E.J., Jenkot, J.L., Keeney, D.B., Lane, R.L., Moyer, G.R., Reading, B.J. & Smith, N.L. 2003. Isolation and characterization of polymorphic microsatellite loci in nurse shark (Ginglymostoma cirratum). Molecular Ecology Notes, 3: 59–61.
Hillis, D.M., Moritz, C. & Mable, B.K. 1996. Molecular Systematics, Second Edition. Sinauer Associates, Inc., Sunderland, MA, USA.
Hoelzel, A.R. 1998. Molecular genetic analysis of populations: A practical approach. Oxford University Press, Oxford, UK.
Hueter, R.E., Heupel, M.R., Heist, E.J. & Keeney, D.B. 2005. Evidence of philopatry in sharks and implications for the management of shark fisheries. Journal of Northwest Atlantic Fishery Science, 35: 239–247.
Keeney, D.B., Heupel, M.R., Hueter, R.E. & Heist, E.J. 2003. Genetic heterogeneity among blacktip shark, Carcharhinus limbatus, continental nurseries along the U.S. Atlantic and Gulf of Mexico. Marine Biology, 143: 1039–1046.
Keeney, D.B. & Heist, E.J. 2003. Characterization of microsalellite loci isolated from the blacktip shark and their utility in requiem and hammerhead sharks. Molecular Ecology Notes, 3: 501–504.
Karl, S.A. & Bowen, B.W. 1992. Global population structure and natural history of the green turtle (Chelonia mydas): RFLP analyses on annonymous nuclear DNA regions. Genetics, 131: 163–173.
Knutsen, H., Jorde, P.E., Andre, C. & Stenseth, N.C. 2003. Fine-scaled geographical population structuring in a highly mobile marine species: the Atlantic cod. Molecular Ecology, 12: 385–394.
Kohler, N.E., Casey, J.G. & Turner, P.A. 1998. NMFS cooperative shark tagging program 1962–93: An atlas of shark tag and recapture data. Marine Fisheries Review, 60: 1–87.
Labate, J.A. 2000. Software for population genetic analyses of molecular marker data. Crop Science, 40: 1521–1528.
Lavery, S. & Shaklee, J.B. 1989. Population genetics of two tropical sharks Carcharhinus tilstoni and C. sorrah, in Northern Australia. Australian Journal of Marine and Freshwater Research, 40: 541–557.
Lewis, P.O. & Zaykin, D. 2001. Genetic Data Analysis: Computer program for the analysis of allelic data. Version 1.0 (d16c). Free program distributed by the authors over the internet from http://lewis.eeb.uconn.edu/lewishome/software.html.
MacDonald, C.M. 1988. Genetic variation, breeding structure and taxonomic status of the gummy shark Mustelus antarcticus in southern Australian waters. Australian Journal of Marine and Freshwater Research, 39: 641–648.
Martin, A.P., Naylor, G.J.P. & Palumbi, S.R. 1992. Rates of mitochondrial DNA evolution in sharks are slow compared with mammals. Nature, 357: 153–155.
Martin, A.P. & Palumbi, S.R. 1993. Protein evolution in different cellular environments: cytochrome b in sharks and mammals. Molecular Biology & Evolution, 10: 873–891.
May, B. 2003. Allozymes. In E.M. Hallerman (ed.). Population genetics: principles and applications for fisheries scientists. American Fisheries Society, Bethesda, MD.
Mcelroy, D., Moran, P., Bermingham, E. & Kornfield, I. 1991. REAP: the restriction enzyme analysis package. Version 4.0. University of Maine, Orono, Maine.
Mills, L.S. & Allendorf, F.W. 1996. The one-migrant-per generation rule in conservation and management. Conservation Biology, 10: 1509–1518.
Murphy, R.W., Sites, J.M.J., Buth, D.G. & Haufler, C.H. 1996. Proteins: isozyme electrophoresis. In D.M. Hillis, C. Moritz & B.K. Mable (eds). Molecular Systematics, pp. 51–120. Sinauer Associates, Inc., Sunderland, MA.
Nei, M. & Tajima, F. 1981. DNA polymorphism detectable by endonucleases. Genetics, 97: 145–163.
O’Connell, M. & Wright, J.M. 1997. Microsatellite DNA in fishes. Reviews in Fish Biology & Fisheries, 7: 331–363.
Palumbi, S.R. & Baker, C.S. 1994. Contrasting population-structure from nuclear intron sequences and mtdna of humpback whales. Molecular Biology and Evolution, 11:426–435.
Pardini, A.T., Jones, C.S., Noble, L.R., Kreiser, B., Malcolm, H., Bruce, B.D., Stevens, J.D., Cliff, G., Scholl, M.C., Francis, M., Duffy, C.A.J. & Martin, A.P. 2001. Sex-biased dispersal of great white sharks - In some respects, these sharks behave more like whales and dolphins than other fish. Nature, 412: 139–140.
Pardini, A.T., Jones, C.S., Scholl, M.C. & Noble, L.R. 2000. Isolation and characterization of dinucleotide microsatellite loci in the Great White Shark, Carcharodon carcharias. Molecular Ecology, 9: 1176–1178.
Raymond, M. & Rousset, F. 1995. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. Journal of Heredity, 86: 248–249.
Rousset, F. 2001. Inferences from spatial population genetics. In D.J. Balding, M. Bishop & C. Cannings (eds). Handbook of statistical genetics, pp. 239–268. John Wiley & Sons, Ltd, New York.
Schneider, S., Roessli, D. & Excoffier, L. 2000. Arlequin ver. 2.000: A software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland.
Schrey, A.W. & Heist, E.J. 2002. Microsatellite markers for the shortfin mako and cross-species amplification in Lamniformes. Conservation Genetics, 3: 459–461.
Schrey, A.W. & Heist, E.J. 2003. Microsatellite analysis of population structure in the shortfin mako (Isurus oxyrinchus). Canadian Journal of Fisheries & Aquatic Sciences, 60(6): 670–675.
Shivji, M., Clarke, S., Pank, M., Natanson, L., Kohler, N. & Stanhope, M. 2002. Genetic identification of pelagic shark body parts for conservation and trade monitoring. Conservation Biology, 16: 1036–1047.
Smith, P.J. 1986. Low genetic variation in sharks (Chondrichthyes). Copeia, 1986: 202–207.
Utter, F.M. 1991. Biochemical genetics and fishery management - an historical - perspective. Journal of Fish Biology, 39: 1–20.
Vos, P., Hogers, R., Bleeker, M., Reijans, M., Vandelee, T., Hornes, M., Frijters, A., Pot, J., Peleman, J., Kuiper, M. & Zabeau, M. 1995. AFLP - a new technique for DNA-fingerprinting. Nucleic Acids Research, 23: 4407–4414.
Waples, R.S. 1998. Separating the wheat from the chaff-patterns of genetic differentiation in high gene flow species. Journal of Heredity, 89: 438–450.
Ward, R.D., Woodwark, M. & Skibinski, D.O.F. 1994. A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. Journal of Fish Biology, 44:213–232.
Weir, B.S. & Cockerham, C.C. 1984. Estimating F-statistics for the analysis of population structure. Evolution, 38: 1358–1370.
Wright, S. 1931. Evolution in Mendelian populations. Genetics, 16: 97–159.
![]()
![]()
![]()