![]()
![]()
![]()
The analyses included in this group, also known as Weende proximate analyses, are applied firstly to materials to be used in formulating a diet as a protein or energy source and to finished feedstuffs, as a control to check that they meet the specifications or requirements established during formulation. These analyses will show the moisture, crude protein (total nitrogen), crude fibre, crude lipids, ash and nitrogen-free extract content of the sample. A fuller description of these analyses can be found in Osborne and Voogt (1978), MAFF (1982) and AOAC (1984).
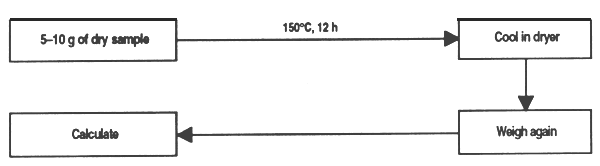
In balancing the ration it is essential to know the water content of each component; also, moisture in prepared feed must be monitored because levels over 8% favour the presence of insects, and over 14% there is the risk of contamination by fungi and bacteria (Cockerell et al., 1971). The method is based on drying a sample in an oven and determining moisture content by the weight difference between dry and wet material.
Apparatus
Method
Calculation
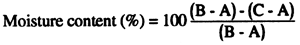
Where:
A = weight of clean, dry scale pan(g)
B = weight of scale pan + wet sample (g)
C = weight of scale pan + dry sample (g)
Figure 1. Determination of humidity content in feed ingredients
Because of its cost this is the most important dietary nutrient in a commercial operation; proper evaluation of it means that the quality of protein intake or of the feed being provided can be controlled. Analysis is by Kjeldahl's method, which evaluates the total nitrogen content of the sample after it has been digested in sulphuric acid with a mercury or selenium catalyst.
Reagents
Material and equipment
Method
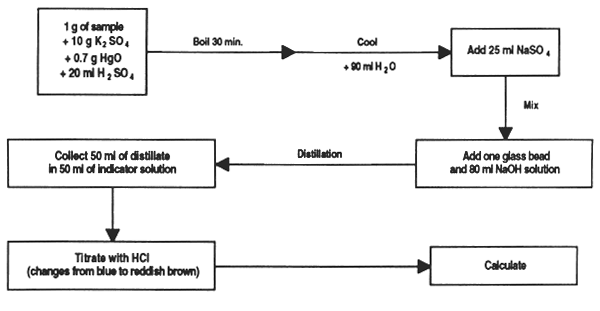
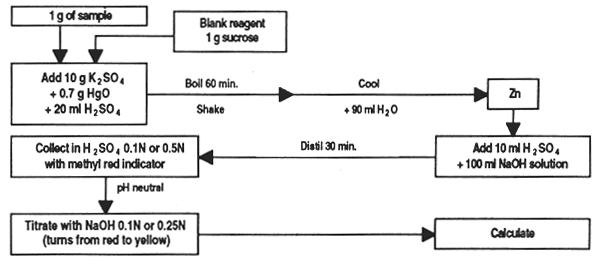
To milligram precision, weigh out 1 g of sample and place in the Kjeldahl flask; add 10g potassium sulphate, 0.7 g mercuric oxide and 20 ml concentrated sulphuric acid.
Place the flask tilted at an angle in the digester, bring to boiling point and retain until the solution is clear; continue to heat 30 minutes more. If foam is too abundant, add a little paraffin wax.
Leave to cool, gradually adding approximately 90 ml distilled, de-ionized water. When cold add 25 ml sodium sulphate solution and stir.
Add one glass bead and 80 ml of 40% sodium hydroxide solution, keeping the flask tilted. Two layers will form.
Quickly connect the flask to the distillation unit, heat and collect 50 ml of distillate containing ammonia in 50 ml of indicator solution.
At the end of distillation, remove the receptor flask, rinse the end of the condenser and titrate the solution with the standard chlorhydric acid solution.
Calculations
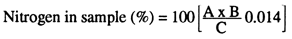
Crude protein (%) = nitrogen in sample × 6.25
Where:
A = chlorhydric acid used in titration (ml)
B = normality of standard acid
C = weight of sample (g)
Figure 2. Determination of crude protein by Kjeldahl's method
Reagents
Material and equipment
Method
Weigh 1 g of sample to within milligrams and transfer it to a Kjeldahl flask; add 10g potassium sulphate or sodium sulphate, 0.6–0.7 g mercuric oxide, 25 ml sulphuric acid and a few grains of pumice stone.
Heat flask moderately at first, shaking occasionally until the material is carbonized and the bubbles have disappeared, then raise the temperature and bring to a gentle boil. Do not allow the flask walls to overheat so that organic particles do not adhere to them.
When the solution looks clear and colourless, continue to boil for 2 hours longer, then cool. If after digestion and cooling the solution crystallizes, repeat the analysis; if crystallization continues to occur, repeat the analysis using more sulphuric acid.
Carefully add 250–350 ml distilled water to the flask, stirring the contents at the same time; let cool and add a few zinc pellets.
Transfer 25 ml sulphuric acid solution 0.1 or 0.5N to the collecting flask of the distilling apparatus according to the anticipated nitrogen value of the sample, and add a few drops of methyl red indicator.
Taking care not to lose ammonia, carefully add to the sample 100 ml sodium hydroxide solution and then 10 ml sodium sulphate solution or 25 ml sodium thiosulphate solution. Mix thoroughly and connect immediately to the distilling apparatus.
Heat the flask so as to distil about 150 ml of the liquid in 30 minutes. Measure the pH of the distillate with litmus paper and if it is alkaline, continue distillation. During this process, stir the contents of the flask occasionally. If the distillate turns alkaline, the determination should be abandoned and the analysis repeated with the appropriate adjustments.
In the collecting flask, titrate the excess sulphuric acid with sodium hydroxide 0.1 or 0.25N, according to the normality of the acid used, to the final point of the methyl red or methyl red-methylene blue indicator.
Run a reagent blank using 1 g of sucrose instead of the sample to use in calculating results.
Calculations
Determine the H2So4 consumed. 1ml acid ≡ 1.4 mg nitrogen.
Calculate the percentage of nitrogen in the sample and convert to percentage of protein by multiplying the result by 6.25.
If the presence of ammonia nitrogen or nitrates is suspected in the sample they must be evaluated to be subtracted from total nitrogen. Except in the case of feed for ruminants, the protein and non-protein nitrogen content must be evaluated and also subtracted from the total nitrogen.
Figure 3. Standard MAFF method for determination of crude protein
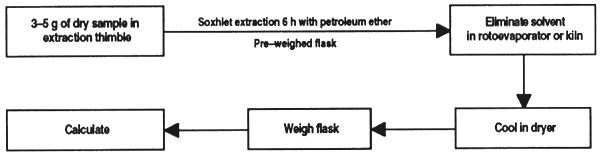
In this method, the fats are extracted from the sample with petroleum ether and evaluated as a percentage of the weight before the solvent is evaporated.
Reagents, material and equipment
Method
Remove extraction flasks from the kiln without touching them with the fingers, cool in a dryer and weigh to within milligrams.
Weigh 3 to 5 g of dry sample to within milligrams in an extraction thimble, handling it with tongs and place in the extraction unit. Connect the flask containing petroleum ether at 2/3 of total volume to the extractor.
Bring to boil and adjust heat to obtain about 10 refluxes per hour. The length of the extraction will depend on the quantity of lipids in the sample. Very fatty materials will take 6 hours.
When finished, evaporate the ether by distillation or in a rotoevaporator. Cool the flasks in a dryer and weigh them to within milligrams. The defatted sample can be used in determining crude fibre.
Calculations
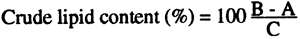
Where:
A = weight of clean dry flask (g)
B = weight of flask with fat (g)
C = weight of sample (g)
Figure 4. Determination of lipids by Soxhlet's method
This method gives the crude fibre content of the sample after it has been digested in sulphuric acid and sodium hydroxide solutions and the residue calcined. The difference in weight after calcination indicates the quantity of fibre present.
Reagents
Sulphuric acid solution 0.255N.
Sodium hydroxide solution 0.313N, free of sodium carbonate.
Antifoam (e.g. octyl alcohol or silicone).
Ethyl alcohol at 95% (v/v).
Petroleum ether.
Chlorhydric acid solution at 1% (v/v).
Material and equipment
600 ml flat-bottomed balloon flask with roughened neck.
Condensation unit for flask.
11 Kitazato flask
Buchner funnel.
Filtration crucible.
Rubber cones.
Whatman No541 filter paper.
500 ml retort.
Dryer.
Laboratory kiln.
Crucible furnace.
Method
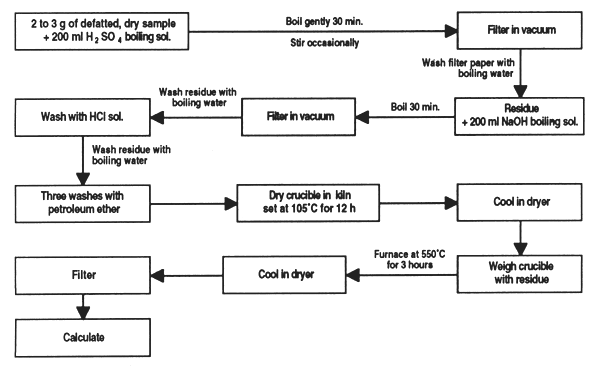
Weigh out 2 to 3 g of defatted, dry sample to within milligrams. Place in the flask and add 200 ml boiling sulphuric acid solution.
Attach the condenser and bring to boiling point in one minute; if necessary, add antifoam. Boil for exactly 30 minutes, maintaining the volume of distilled water constant and swirling the flask periodically to remove particles adhering to the sides.
Line the Buchner funnel with the filter paper and pre-heat with boiling water. At the same time, at the end of the boiling period, remove the flask, let rest one minute and filter the contents carefully, using suction. Filtration should be carried out in less than 10 minutes. Wash the filter paper with boiling water.
Transfer the residue to the flask using a retort containing 200 ml of boiling NaOH solution and boil for 30 min. as in step (ii).
Preheat the filtration crucible with boiling water and carefully filter the hydrolyzed mixture after letting it rest for 1 min.
Wash the residue with boiling water, with the HCI solution and then again with boiling water, finishing with three washes with petroleum ether. Place the crucible in a kiln set at 105°C for 12 hours then cool in dryer.
Quickly weigh the crucible with the residue inside (do not handle them) and place in the crucible furnace at 550° C for 3 hours. Leave to cool in a dryer and weigh them again.
Calculations
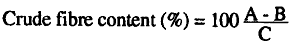
Where:
A = weight of crucible with dry residue (g)
B = weight of crucible with ash (g)
C = weight of sample (g)
Recommendations
One of the problems most often encountered in evaluating crude fibre is filter blockage, this is why in some cases it is recommended to substitute a piece of cotton fabric for filter paper [step (iv)]. To avoid saturation of the filter crucible [step (vi)], tilt it slightly and add the material to be filtered very slowly so that it covers the filtering surface little by little.
Filtration crucibles tend to block up with use. To clean, calcine them at 500°C and force water through in reverse. When they are blocked with mineral particles, prepare a solution containing 20% KOH, 5% Na3PO4 and 0.5% EDTA sodium salt; heat and force through the crucible in reverse. This treatment erodes the glass filter.
Figure 5. Determination of crude fiber
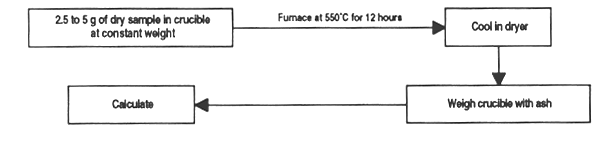
This method is used to determine ash content in feedstuffs by calcination. Ash is considered as the total mineral or inorganic content of the sample.
Material and equipment
Method
Place 2.5 to 5 g of dry sample in a crucible previously calcined and brought to constant weight.
Place the crucible in a furnace and heat at 550°C for 12 hours; leave to cool and transfer to a dryer.
Carefully weigh the crucible again with the ash.
Calculations
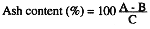
Where:
A = weight of crucible with sample (g)
B = weight of crucible with ash (g)
C = weight of sample (g)
Figure 6. Determination of ash content in feed ingredients
This includes all the nutrients not assessed by the prior methods of proximate analysis. These are composed mainly of digestible carbohydrates, vitamins and other non-nitrogen soluble organic compounds. Since the result is obtained by subtracting the percentages calculated for each nutrient from 100, any errors in evaluation will be reflected in the final calculation.
Calculations
Nitrogen-free extract (%) = 100 - (A + B + C + D + E)
Where:
A = humidity content (%)
B = crude protein content (%)
C = crude lipid content (%)
D = crude fibre content (%)
E = ash content (%)
Because analyses are usually conducted with specially prepared samples, certain corrections must be made to the results so that they reflect the actual nutrient content of the material under the conditions in which it will be used.
Moisture
If the analyses were made on a dry basis (DB), i.e with dehydrated material the result must be corrected to express it on a moist basis (MB) as found in the feed or material for it, using the following formula:
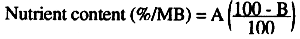
Where:
A = nutrient content (%DB)
B = moisture content of material (%)
Lipids
When defatted material is used, for example in the analysis for crude fibre, a similar formula is applied to obtain a value representative of the sample:

Where:
A = fibre content (defatted, %)
B = lipid content of the material (%)
![]()
![]()
![]()