![]()
![]()
![]()
The nutritional value of any feed depends largely on the quality of ingredients used. This is measured in terms of their capacity to encourage growth and maintain the animals in good condition. Therefore, the origin of materials and/or the prior treatment to which they are subjected influence their quality as far as the availability of nutrients that can be used in the animal's metabolic functions. They can also affect the content of endogenous toxic factors characteristic of each material, particulary those of vegetable origin, that act as anti-nutrients and adversely affect the organism.
There are several techniques for finding out the availability of a particular nutrient, mainly amino-acids, and also for determining the presence of toxic substances so that they can either be eliminated or the material rejected. This document contains the methods for analyzing the anti-nutrients most commonly found in plant protein.
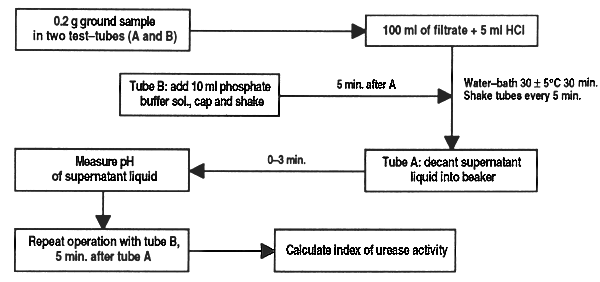
This method of analysis determines residual urease in soyabean meal and its by-products according to the Caskey-Knapp method (1944) modified by AACC (1969) and Leticia Comar (Nutrimentos del Sureste, Merida, Yucatan, Mexico). The result is given in pH units proportional to urease activity. Acceptable values fluctuate between 0.05 and 0.5; lower values indicate overcooking, and higher values, undercooking.
Reagents
Material and equipment
Method
Weigh double samples of 0.2 g finely ground (without overheating) soyabean meal and transfer to test-tubes (A and B); to tube A add 10 ml of urea buffer solution. Stopper, shake and place in water bath for 30 min at 30 ± 0.5°C.
After 5 min, add to tube B 10 ml phosphate buffer solution, stopper, shake and place in water bath.
Shake each tube every 5 minutes (6 times in 30 min).
Remove tube A from water bath and decant supernatant liquid into a 10 ml beaker and measure pH in under 3 min. Repeat process with tube B 5 min after tube A.
Calculate the index of urease activity as pH units resulting from the difference between readings of tube A and tube B and determine sample quality according to the table below:
IAU = pH tube A (sample + urea buffer) - pH tube B (sample + phosphate buffer)
| Degree of baking | pH units | pH tube A | pH tube B | Colour |
| Raw | 1.90 | 8.80 | 6.90 | Red |
| Underbaked | 0.80 | 7.60 | 6.80 | Pink |
| Properly baked | 0.30 | 7.10 | 6.80 | Pink |
| Properly baked | 0.08 | 6.98 | 6.90 | Pale pink |
| Overbaked | 0.03 | 6.93 | 6.90 | Amber |
Figure 24. Determination of urease activity in soyabean meal and its derivatives
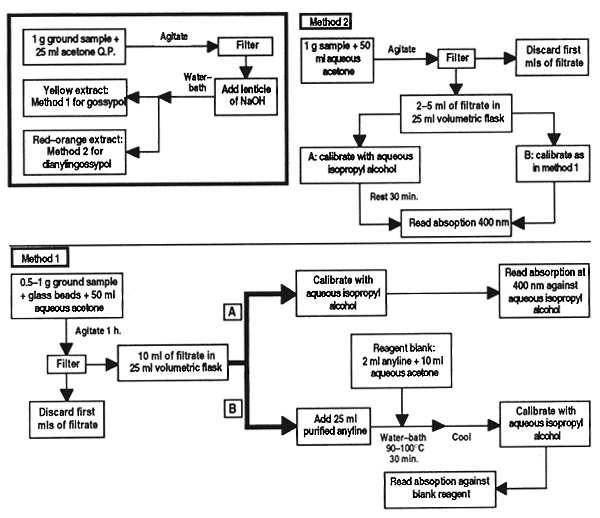
This anti-nutritional factor is a typical feature of cottonseed meal. Content of it must be determined because of its detrimental effects on animals fed with it, even though fish appear to support higher concentrations of the alkaloid due to the high protein content of their diet. This paper describes two methods, the first for normal meal and the second for chemically treated meals that contain di-aniline gossypol.
Reagents
Material and equipment
Method
Grind the sample so that it will pass through a 1 mm screen, taking care not to overheat. Take approximately 1 g of the sample and add 25 ml pure acetone. Shake for a few minutes, filter and divide the filtrate into two parts. To one part add a pellet of sodium hydroxide and heat in a water bath for a few minutes. A yellowish extract that does not change colour with the hydroxide indicates that the meal has not been chemically treated, so proceed with method 1. If a deep orange-red colour appears it indicates the presence of di-anilinegossypol and method 2 must be followed.
Method 1
Weigh 0.5–1 g of sample, depending on the amount of gossypol anticipated, into a Erlenmeyer flask and add 2 or 3 glass beads. Add 50 ml aqueous acetone solution, stopper the flask and agitate for on e hour. Filter, discard the first ml of filtrate and then take two equal parts (2–10 ml, depending on the gossypol content expected) and transfer to 25 ml volumetric flasks. Dilute one of the parts, calibrate to volume with aqueous iso-propyl alcohol [solution (a)]. To the other part [solution (b)] add 2 ml purified aniline, heat in water bath (90–100°C) for 30 min together with a blank containing 2 ml aniline and a volume of aqueous acetone solution equal to the aliquot. Remove solution b and the blank, add sufficient aqueous iso-propyl alcohol to make a monogeneous solution and cool to room temperature in a bath of water. Calibrate with aqueous iso-propyl alcohol.
Read the samples in the spectrophotometer at an absorption of 400 nm. Adjust the instrument to 0 obsorption with aqueous iso-propyl alcohol and determine the absorption of solution (b) and the blank reagent. If the blank is below 0.022 absorption, continue with the analysis; if not, repeat using freshly distilled aniline.
Determine the absorption of solution (b) with the blank reagent; adjust to 0 absorption. Calculate the corrected absorption of the aliquot: absorption of solution (b) minus absorption of solution (a). Determine the mg of free gossypol present in the solution of sample using the calibration curve.
Method 2
Weigh 1 g of sample in a Erlenmeyer flask, add 50 ml aqueous acetone, shake and filter as above. Add duplicate aliquots of the filtrate (2 to 5 ml, depending on the amount of free gossypol expected) to 25 ml volumetric flasks. Calibrate one of the samples [solution (a)] with aqueous iso-propyl alcohol and leave for 30 min before reading it on the spectrophotometer. Calibrate the other aliquot [solution (b)] as in method (1), determine the absorption of solutions (a) and (b) as in the method above and calculate the apparent content of gossypol in the two solutions, using the calibration curve.
Preparation of calibration curve
Take duplicate samples of 1, 2, 3, 4, 5, 7, 8 and 10 ml of standard 0.02 mg/ml gossypol standard and transfer to 25 ml volumetric flasks. Dilute one series [solution (a)] calibrating with aqueous iso-propyl alcohol and determine absorption as above. To the other series [solution (b)] add 2 ml purified aniline and proceed as above. Prepare a blank reagent with 2 ml aniline and 10 ml aqueous acetone, heated together with the standard. Determine absorption as in method 1 and calculate the optical density corrected for each standard:
Corrected absorption = Abs. sol (b) - Abs. sol (a)
Draw the standard curve, plotting corrected absorption against gossypol concentration in 25 ml.
Calculation
Calculate % of free gossypol in normal meals:
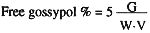
| Where: | G = graph reading |
| W = weight of sample | |
| V = volume of aliquot used. |
For chemically treated meals:
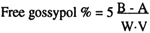
| Where: | A = mg of free gossypol appearing in aliquot (a) |
| B = mg of free gossypol appearing in aliquot (b). |
Figure 25. Determination of free gossypol in cottonseed meal
The method described shows the approximate content of thioglucides but does not determine individual thioglucides and isocyanates.
Reagents and apparatus
Method
To 10 g of meal (defatted by Soxhlet extraction) add 250 ml distilled water, hydrolyze at 54°C for 1 h and heat at boiling point for 2 h, maintaining volume constant.
Filter, reserving the filtrate, and wash the residue three times with 50 ml hot water, adding the liquid to the initial filtrate, and calibrate to 600 ml.
Precipitate the barium sulphate by heating and adding abundant barium chloride solution. Leave in a vapour bath for a few hours then filter.
Calcine in a crucible furnace and weigh the precipitate.
Calculation
Calculate the approximate thioglucide content thus:
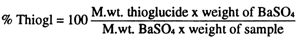
Figure 26. Determination of thioglucides in feed ingredients
This method is suitable for materials such as groundnut meal, copra meal and palm kernel meal. For further details and alternative procedures see Methods of Aflatoxin Analysis by B.D. Jones (1972), Report NoG70, Tropical Products Institute, London.
Reagents
Material and equipment
Method
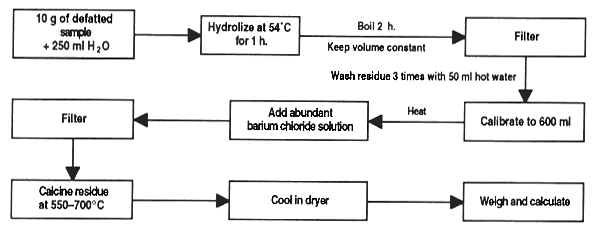
Weigh 10 mg of the material into a wide-mouthed flask and mix with 10 ml water (if a material high in fat is being used, a prior Soxhlet extraction will be necessary). Add 100 ml chloroform and agitate for 30 min.
Filter the mixture through the cellulite, take 20 ml of the filtrate and bring to 25 ml [solution (a)]. Take another 20 ml of filtrate and concentrate to 5 ml [solution (b)].
Prepare chromatography plaques by agitating 100 g “G” silica gel with 220 ml water for 20 min and applying the mix to the plates with the proper means, at a thickness of 509 μ. Leave to rest for 1 hour and dry at 100°C.
Drop 10 and 20 μl of solution (b) and 15 and 10 μl of solution (b) on the plaque together with one drop of standard qualitative solution leaving 2 cm at the bottom of the plate and 2 cm at each side. Do this under faint light.
Reveal the plate in ethyl ether to a height of 12 cm. Let dry in soft light and then reveal again in the chloroform/methanol mixture to a height of 10 cm over the base line.
Examine the plate in a dark room at 30 cm from the UV source. A fluorescent blue stain at Rf 0.5–0.55 indicates aflatoxin (check that the stain of the standard also falls within this range). The presence of another stain at Rf 0.45–0.5 denotes aflatoxin G.
Figure 27. Chromatographic determination of aflatoxins in feed ingredients
The toxicity of a sample can be classified in terms of aflatoxins B and G according to the following table:
| COMPARATIVE TABLE FOR DETERMINING AFLATOXIN B AND G TOXICITY | |||
| Volume applied (ml) | Conc. of aflatoxin (mg/kg) | Toxicity of observed fluorescence | |
| no fluorescence | w/fluorescence | ||
| 5 [sol. (a)] | < 1,000 | > 1,000 | Very high |
| 10 [sol. (a)] | < 500 | 500 – 1,000 | High |
| 10 [sol. (b)] | < 100 | 100 – 500 | Medium |
| 20 [sol. (b)] | < 50 | 50 – 100 | Low |
After Harris, 1980.
Mimosine is a free amino-acid very often present in certain legumes which include Leucaena leucocephala and L. glauca, both considered as excellent sources of protein for animal feed. Its presence limits the use of the leaves and seeds in feed for monogastric animals since it affects thyroid function, leading to poor growth.
Reagents
Material and equipment
Method
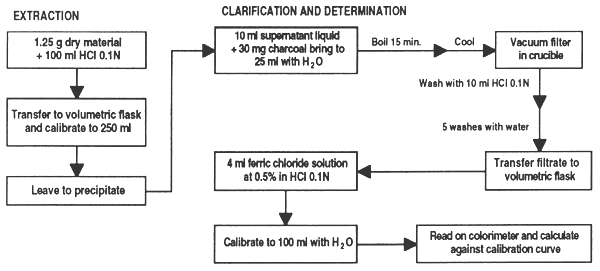
Extraction: place 1.25 g dry leucaena (kathin) leaf in a 250 ml beaker and digest with HCl 0.1N for one hour, agitating constantly, let cool and transfer all to a 250 ml volumetric flask. Shake well and leave until the solids settle on the bottom.
Clarification: transfer 10 ml of supernatant liquid to a 150 ml beaker with 30 mg charcoal. Add water to bring the volume up to 25 ml, cover with a watchglass and bring to boil for 15 min. Leave to cool and filter under vacuum in the filtration crucible; the filtrate is collected in a test-tube placed in the Kitazato flask. Rinse the beaker and the material in the crucible with 10 ml HCl 0.1N divided into three parts. Finally rinse the crucible with 5 small parts of water.
Colorimetric determination: transfer the liquid obtained to a 100 ml volumetric flask, add 4 ml ferric chloride solution at 0.5% in HCl 0.1N and calibrate with water. The pH should be between 1.5 and 2.5. Read on the colorimeter and calculate the quantity of mimosine in a calibration curve.
Calibration curve: dissolve 0.1046 g pure mimosine in HCl 0.1N and calibrate to 100 ml with the acid. The solution should contain 0.1% mimosine. Put 1 ml of the mimosine solution in a 100 ml volumetric flask and add 10 ml HCl 0.1N and 4 ml ferric chloride solution at 0.5% and calibrate with water. Read the colour intensity on the colorimeter (spectrum 535 nm). Use distilled water as reference and repeat the process using 2, 3, 4 and 5 ml mimosine solution. A line is obtained by plotting the colorimeter reading against concentration.
Figure 28. Colorimetric determination of mimosine in feed ingredients
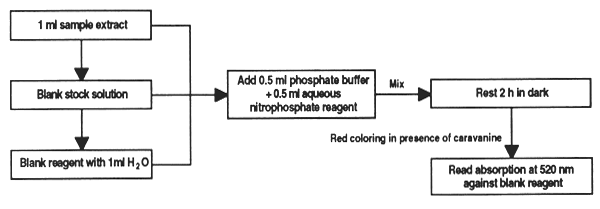
The seeds and leaves of legumes, particularly those of canavalia (Canavalia ensiformis) and sesbania (Sesbania sp.) contain variable amounts of the free amino-acid canavanine, which is toxic for monogastric animals. With this method the amount of this anti-nutrient in feed ingredients can be determined.
Reagents
Phosphate buffer 1 M, pH 7.2. Mix 34.5 g NaH2PO4.H2O with 130.6 g K2PO4 and adjust the volume to 1 litre.
Solution of sodium nitroprussiate 2%. A recently prepared solution should be used. It is stable for one week when stored in darkness at 0–4°C.
Hydrogen peroxide (30% H2O2).
Potassium carbonate solution at 20%.
Stock solution of canavanine. 2.5 mg canavanine/ml. Store in dry ice.
Standard solution of canavanine. Bring 1 ml stock solution to 10ml with water.
Aqueous prussiate reagent. Mix 0.5 ml of 20% carbonate solution and 0.4 ml H2O2 with 10 ml nitroprussiate solution. Let rest 30 min at room temperature (20–25°C). Must be prepared daily. Larger quantities of peroxide will lighten the colour obtained with the canavanine.
Material and equipment
Method
Place 1ml of sample extract containing 0–025 mg canavanine in a test-tube; add 0.5 ml phosphate buffer and 0.5 ml aqueous prussiate reagent. In the same way prepare blanks containing 0.2, 0.5, 0.8 and 1 ml diluted stock solution and bring to 1 ml with water. Also prepare a reagent blank with 1ml water, treated with the phosphate and aqueous prussiate.
Stir vigorously and store tubes in a dark cabinet; a reddish colour will develop in the presence of canavanine.
After 2 hours, read in a colorimeter or spectrophotometer. In the latter, adjust the wavelength to 520 nm and the blank to zero optical density. Spectrophotometer cells can be used to develop the colour; if cells over 2 ml are used, up to 10 ml water can be added to them after the 2 hours of incubation to develop the colour.
Calculations
Plot the optical density of each standard against the corresponding quantity of canavanine; the canavanine content of the samples is calculated on the basis of the curve.
Figure 29. Colorimetric determination of canavanine in feed ingredients
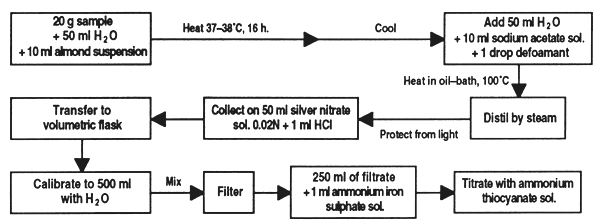
Chlorhydric acid is naturally present in different materials such as linseed, cassava meal and some legume seeds. This method can be used to determine the free acid or that present in the form of glucosides. For this, the sample is suspended in water, the acid is freed by means of enzymes and separated by distillation to be collected in an acidified solution of silver nitrate.
Reagents
Material and equipment
Method
Weigh 20 g of prepared sample to within 0.005 g, place in a 1,000 ml flat-bottomed flask, add 50 ml water and 10 ml suspension of sweet almonds. Stopper the flask and place in kiln for 16 hours at 37–38°C. Cool to room temperature, add 80 ml water, 10 ml sodium acetate solution and one drop of antifoam.
Connect the flask to the distillation apparatus and place in an oil bath previously adjusted to slightly over 100°C; distil 200–300 ml of liquid by passing a current of steam through the flask and gently heating the oil bath. Collect the distillate in a Erlenmeyer flask protected from light containing exactly 50 ml 0.02N silver nitrate solution and 1 ml nitric acid. Make sure that the condenser extension is submerged in the silver nitrate solution
Transfer the contents of the Erlenmeyer flask to a 500 ml volumetric flask and bring to volume with water, stir and filter. Remove 250 ml of filtrate, add approximately 1 ml ammoniated ferric sulphate and titrate excess silver nitrate with the ammonia thiocyanate solution. If wished, a blank test can be run following the same method with 10ml sweet almond suspension omitting the sample.
Expression of results
If the blank shows that the silver nitrate solution has been consumed, subtract its value from the volume consumed by distillation of the sample. 1 ml of 0.02N AgNO3 = 0.54 mg HCN. Express the result as a percentage of the sample.
NOTE:
If the sample (e.g. beans) has a high sulphur content, a black precipitate of silver sulphate will form; this is filtered together with the silver cyanide deposit. The formation of this precipitate consumes silver nitrate solution and the effect must be eliminated by calculating the HCN content. To do this, treat the deposit on the filter paper with 50 ml ammonia to dissolve the silver cyanide. Wash the residue in dilute ammonia and determine the silver content. Convert the value to ml of solution 0.02N of silver nitrate solution and subtract from the volume of silver nitrate solution consumed by the sample distillate.
Figure 30. Determination of chlorhydric acid in feed ingredients
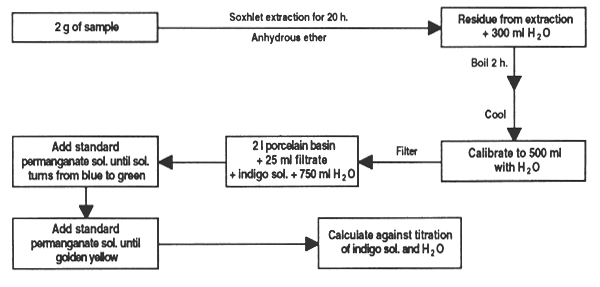
Tannins are water-soluble polypeptide glucosides present in many plants; their sourness and astringency give feed an unpleasant taste and odour. They affect protein utilization because they bind to lysine and so make it unavailable to monogastric animals.
Reagents
Method
Extract 2 g of sample with anhydrous ether for 20 hours. Boil the residue for 2 hours with 300 ml water; cool, dilute to 500 ml and filter.
Measure 25 ml of the infusion into a 21 porcelain bowl, add 20 ml indigo solution and 750 ml water. Add the standard permanganate solution, 1 ml at a time until the colour changes from blue to green; than add a few more drops until it becomes golden yellow.
Similarly, titrate a mixture of 20 ml indigo solution and 750 ml water. Multiply the difference between titrations by the desired factor to obtain quercitannic acid or absorbed oxygen.
Figure 31. Determination of tannins in feed ingredients
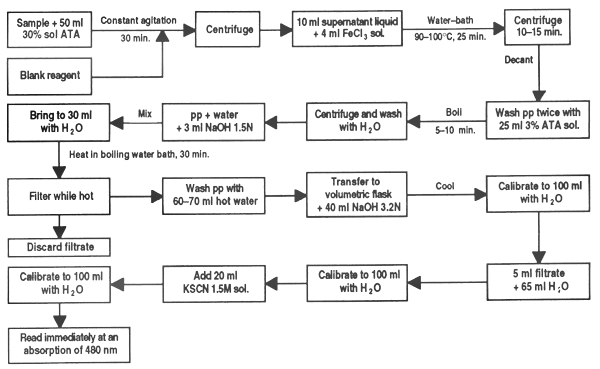
Phytic acid may be found in certain grains such as sesame, wheat, rice, oats, sorghum, barley, etc. Its presence can cause phosphorous deficiency in monogastric animals by binding and forming indigestible phytates that make this element unavailable. It also affects the availability of calcium, magnesium or the iron used to form the phytin complex.
Reagents
Material and equipment
Method
In a 125 ml Erlenmeyer flask place a finely ground sample (40 screen) estimated to contain 5 to 50 g P-phytate. Extract for 30 min with 50 ml 3% solution of TAA with constant agitation.
Centrifuge the suspension and transfer a 10 ml aliquot of the supernatant liquid to a conical centrifuge tube. Add 4 ml FeCl3 solution prepared to contain 2 mg Fe2, blowing through the pipette rapidly.
Heat the tube containing the sample for 45 min in a water bath at 90–100°C. If the supranatant is not clear after 30 min, add 1 or 2 drops of the solution of sodium sulphate at 3% in TAA and continue to heat. Centrifuge for 10–15 min and carefully pour off the supranatant liquid. Wash the precipitate twice with 20–25 ml TAA 3% solution, dispersing it well and heating for 5–10 min in boiling water; centrifuge. Finally, repeat the wash once with water.
Disperse the precipitate in a little distilled water, add 3 ml of 1.5N NaOH solution and stir. Bring the volume to approximately 30 ml with distilled water and heat in boiling water for 30 min. Filter while hot (quantatively) with a moderately retaining paper (Whatman No2 or SyS 597).
Wash the precipitate with 60–70 ml hot distilled water and discard the filtrate. The filtrate can be used if a determination of phosphate is required. Dissolve the precipitate left in the paper with 40 ml of the 3.2N solution of HNO3 transferring it to a 100 ml volumetric flask. Wash the paper several times with distilled water, collecting it in the flask. Cool the sample to room temperature and calibrate with distilled water.
Transfer a 5 ml sample to a 100 ml volumetric flask and dilute to approximately 70 ml. Add 20 ml 1.5M KSCN solution and calibrate with distilled water. Immediately (within 1 min) read in the spectrophotometer at 480 nm.
Run a blank reagent with each sample. Calculate the iron content on the basis of a Fe(NO3) standard run at the same time or with a standard curve prepared earlier.
Calculation
Calculate the P-phytate from the iron results, assuming a molecular rate of 4:6 iron: phosphor.
Figure 32. Determination of phytic acid in feed ingredients
Saponins can be found in a wide variety of plants including alfalfa, soya beans and legume seeds and have shown negative effects on the growth of monogastric animals. This anti-nutrient is heat stable.
Reagents
Material and equipment
Method
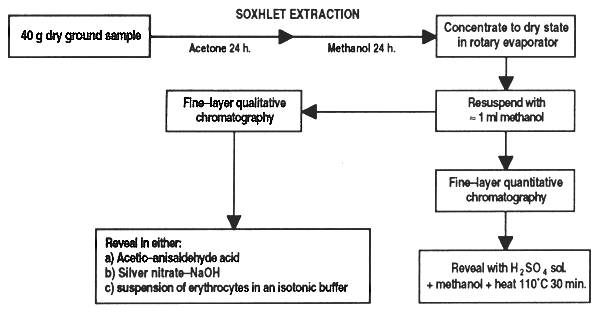
Grind the sample finely and dry at constant weight. Weigh out about 40 g and place in the Soxhlet reflux extractor with acetone for at least 24 hours to remove lipids, pigments, etc… Change the solvent for methanol and continue extraction for at least another 24 hours. Let the methanolic extract cool and make up to 250 ml with methanol.
At this point there is a modification to the method, proposed by Miriam Monforte (CICY, Merida, Yucatan, Mexico). Instead of bringing the sample up to 250 ml as suggested in the original method it is more recommendable to concentrate it, which makes it possible to quantify very small amounts of saponin. To do this, transfer the methanolic extract into a rotary evaporator and concentrate until dry. Suspend the residue again in a minimum of methanol and transfer to a preweighed vial; if it is not to be used immediately, dry with nitrogen and store in the dryer. Weigh the vial with the dry sample and calculate the weight of the residue. To use the sample, suspend again in a known quantity of methanol (around 1 ml).
Qualitative fine-layer chromatography: place fine drops of the extract on a silica gel plaque and reveal with a solution of concentrated n-buthanol-ethanol-ammonia solution (3.5:1:2.5). Three different methods can be used to identify saponin stains: (i) aspersion with acetic-anisaldehyde acid; (ii) treatment with silver nitrate followed by sodium hydroxide, and (iii) treatment with a suspension of erythrocytes in an isotonic buffer. For further details see Fenwick and Oakenfull (1981).
Quantitative fine-layer chromatography: place a series of fine drops of a standard solution of saponin on the chromatography plates. These points of extract should be placed so that each one is at the side of standard saponin drops. Reveal the plates in the same way as described above; the drops should be visualized with aspersion with a solution of sulphuric acid in methanol, followed by heating at 110°C for 30 min. Measure the intensity of the saponin stains with a densitometer and calculate the peak areas on the plotter with a planimater. The results are expressed as the relation (R) of the peak areas of the unknown sample in respect to those of the standard.
It has been shown that the saponin concentration in the sample (in μg) is proportional to the square of relative areas (R2). Plot R2 against the volume of the drop of methanolized extract on the plate. The downslope of the line (calculated by the least squares method), divided by the gradient of a line derived from a master curve, will give the concentration of saponin in the extract and thus the saponin content of the sample. This method may have a cumulate experimental error of ±20%.
Figure 33. Determination of saponine in feed ingredients
![]()
![]()
![]()