

ALINORM04/27/23
APPENDIX V
PROPOSED DRAFT GUIDELINES FOR EVALUATING ACCEPTABLE
METHODS OF ANALYSIS
(At Step 5 of the Procedure)
1. These guidelines provide a framework for evaluating acceptable methods of analysis.
2. These guidelines are intended to assist countries in the application of requirements for trade in foodstuffs in order to protect the consumer and to facilitate fair trade.
3. Laboratories involved in the evaluation should comply with Codex Guidelines CAC/GL 27 on the competence of testing laboratories involved in the import and export of foods.
4. If a method of analysis has been endorsed by Codex, then preference should be given to using that procedure.
5. Methods should be assessed as appropriate against the following criteria by laboratories involved in the import and export control of foods:
• accuracy
• applicability (matrix, concentration range and preference given to 'general' methods)
• detection/determination limits
• linearity
• precision; repeatability intra-laboratory reproducibility inter-laboratory
• recovery
• selectivity (interference effects etc.)
• sensitivity
6. Their definition and approach to their estimation are given below.
Definition
(as a concept)
The closeness of agreement between the reported result and the accepted reference value.
Note:
The term accuracy, when applied to a set of test results, involves a combination of random components and a common systematic error or bias component. {ISO 3534-1} When the systematic error component must be arrived at by a process that includes random error, the random error component is increased by propagation of error considerations and is reduced by replication.
(as a statistic)
The closeness of agreement between a reported result and the accepted reference value. {ISO 3534-1}
Note:
Accuracy as a statistic applies to the single reported final test result; accuracy as a concept applies to single, replicate, or averaged value.
Estimation
Wherever possible the use of traceable reference materials (matrix matched and similar level of analyte) should be used to determine the accuracy of the method of analysis used.
NMKL Procedure 9 (2001)If certified reference materials are used during a method evaluation exercise then the mean determined value can be compared against the mean known value by calculation of the z-value.
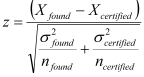
or, if certified reference material standard deviation data are unavailable 95% confidence limit data may be used as an estimate of certified reference material standard deviation.
If the reference value is X certified ±CI (95% confidence interval) then:
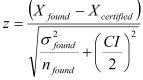
A z-value outside the range |z|≤2 indicates a significant bias and a bias correction should be made.
APPLICABILITY
The analytes, matrices, and concentrations for which a method of analysis may be used satisfactorily to determine compliance with a Codex standard.
Note:
In addition to a statement of the range of capability of satisfactory performance for each factor, the statement of applicability (scope) may also include warnings as to known interference by other analytes, or inapplicability to certain matrices and situations.
Estimation
This should detail the analytes, matrices and concentrations for which the method of analysis may be used satisfactorily to determine compliance with a Codex standard. This may also include warnings as to known interference by other analytes, or inapplicability to certain matrices and situations. The Youden approach a fractional factorial approach, is commonly used to assess applicability/ruggedness.
DETECTION/DETERMINATION LIMITS
Definition: Detection Limit
The detection limit is conventionally defined as field blank + 3σ, where σ is the standard deviation of the field blank value signal (IUPAC definition).
However, an alternative definition which overcomes most of the objections to the above approach (i.e. the high variability at the limit of measurement can never be overcome) is to base it on the rounded value of the reproducibility relative standard deviation when it goes out of control (where 3 σR = 100%; σR = 33%, rounded to 50% because of the high variability). Such a value is directly related to the analyte and to the measurement system and is not based on the local measurement system.
Definition: Determination Limit
As for detection limit except that 6σ or 10σ is required rather than 3σ.
However, an alternative definition that corresponds to that proposed for the detection limit is to use σR = 25%. This value does not differ much from that assigned to the detection limit because the upper limit of the detection limit merges indistinguishably into the lower limit of the determination limit.
Estimation
Where measurements are made at low analyte or property levels, e.g. in trace analysis, it is important to know what is the lowest concentration of the analyte or property value that can be confidently detected by the method. The importance in determining this, and the problems associated with it, arise from the fact that the probability of detection does not suddenly change from zero to unity as some threshold is crossed. The problems have been investigated statistically in some detail and a range of decision criteria proposed.
For validation purposes it is normally sufficient to provide an indication of the level at which detection becomes problematic. For this purpose the “blank + 3s” approach will usually suffice. Where the work is in support of regulatory or specification compliance, a more exact approach such as that described by IUPAC and various others is likely to be appropriate. It is recommended that users quote whichever convention they have used when stating a detection limit.
Detection Limit - Quick Reference | |
What to analyse |
What to calculate from the data |
a) 10 independent sample blanks measured once each. |
Sample standard deviation ‘s’ of a) sample blank values, or b) fortified sample blank values |
or |
|
b) 10 independent sample blanks fortified at lowest acceptable concentration measured once each |
Express Detection Limit as the analyte concentration corresponding to a) mean sample blank value + 3s or b) 0 + 3s |
This approach assumes that a signal more than 3s above the sample blank value could only have arisen from the blank much less than 1% of the time, and therefore is likely to have arisen from something else, such as the measurand. Approach a) is only useful where the sample blank gives a non-zero standard deviation. Getting a true sample blank can be difficult. | |
c) 10 independent sample blanks fortified at lowest acceptable concentration, measured once each |
Sample standard deviation ‘s’ of the fortified sample blank values |
Express Detection Limit as the analyte concentration corresponding to sample blank value +4.65s
| |
The ‘lowest acceptable concentration’ is taken to be the lowest concentration for which an acceptable degree of uncertainty can be achieved.
| |
The determination limit is strictly the lowest concentration of analyte that can be determined with an acceptable level of repeatability precision and trueness. It is also defined by various conventions to be the analyte concentration corresponding to the sample blank value plus 6 or 10 standard deviations of the blank mean.
Note: Neither Detection Limit nor Determination Limit represent levels at which quantitation is impossible. It is simply that the size of the associated uncertainties approach comparability with the actual result in the region of the Detection Limit.
Determination Limit– Quick Reference | |
What to analyse |
What to calculate from the data |
a) 10 independent sample blanks measured once each. |
Sample standard deviation ‘s’ of sample blank value.
|
Getting a true sample blank can be difficult. |
|
b) Fortify aliquots of a sample blank at various analyte concentrations close to the Detection Limit. |
Calculate the standard deviation ‘s’ of the analyte value at each concentration. Plot s against concentration and put assign a value to the Determination Limit by inspection. |
Measure, once each, 10 independent replicates at each concentration level. |
Express Determination Limit as the lowest analyte concentration which can be determined with an acceptable level of uncertainty. |
Normally Determination Limit forms part of the study to determine working range. It should not be determined by extrapolation below the lowest concentration fortified blank. | |
If measurements are made under repeatability conditions, a measure of the repeatability precision at this concentration is also obtained. | |
LINEARITY
Definition
The ability of a method of analysis, within a certain range, to provide an instrumental response or results proportional to the quantity of analyte to be determined in the laboratory sample. This proportionality is expressed by an a priori defined mathematical expression. The linearity limits are the experimental limits of concentrations between which a linear calibration model can be applied with a known confidence level (generally taken to be equal to 1%).”
Estimation
For any quantitative method, it is necessary to determine the range of analyte concentrations or property values over which the method may be applied. Note this refers to the range of concentrations or property values in the solutions actually measured rather than in the original samples. At the lower end of the concentration range the limiting factors are the values of the limits of detection and/or quantitation. At the upper end of the concentration range limitations will be imposed by various effects depending on the instrument response system.
Within the working range there may exist a linear response range. Within the linear range signal response will have a linear relationship to analyte concentration or property value. The extent of this range may be established during the evaluation of the working range. Note that regression calculations on their own are insufficient to establish linearity. To do this a visual inspection of the line and residuals may be sufficient; objective tests, such as ‘goodness-of-fit’ tests, are better still. In general linearity checks require points at at least 10 different concentrations/property values.
Evaluation of the working and linear ranges will also be useful for planning what degree of calibration is required when using the method on a day-to-day basis. It is advisable to investigate the variance across the working range Within the linear range, one calibration point may be sufficient, to establish the slope of the calibration line. Elsewhere in the working range, multi-point (preferably 6+) calibration will be necessary. The relationship of instrument response to concentration does not have to be perfectly linear for a method to be effective but the curve should be repeatable from day to day. Note that the working and linear range may be different for different matrices according to the effect of interferences arising from the matrix.
Working and Linear Range - Quick Reference | |||
Analyse |
Repeats |
What to calculate from the data |
Comments |
1. Blank plus reference materials or fortified sample blanks at various concentrations |
1 |
Plot measurement response (y axis) against measurand concentration (x axis).
|
Ideally the different concentrations should be prepared independently, and not from aliquots of the same master solution. |
Need at least 6 concentrations plus blank |
Then go to 2. |
This will give visual confirmation of whether or not the working range is linear. This stage is necessary to test a working range, thought to be linear and where it is intended to use single point calibration. | |
2. Reference materials or fortified sample blanks at at least 6 different concentrations within the linear range |
3 |
Plot measurement response (y axis against measurand concentration (x axis). Visually examine for outliers that may not be reflected in the regression.
|
It is unsafe to remove outliers without first checking using further determinations at nearby concentrations.
|
Then go to 3. |
|||
3. As for Determination Limit (b) |
As for Determination Limit.
|
Work with successively lower concentrations until the accuracy and precision becomes unacceptable. | |
PRECISION CHARACTERISTICS
Definitions
The closeness of agreement between independent test results obtained under stipulated conditions {ISO 3534-1}
Notes: {ISO 3534-1}
1. Precision depends only on the distribution of random errors and does not relate to the true value or to the specified value.
1. The measure of precision is usually expressed in terms of imprecision and computed as a standard deviation of the test results. Less precision is reflected by a larger standard deviation.
1. “Independent test results” means results obtained in a manner not influenced by any previous result on the same or similar test object. Quantitative measures of precision depend critically on the stipulated conditions. Repeatability and reproducibility conditions are particular sets of extreme conditions.
Repeatability [Reproducibility]: Precision under repeatability [reproducibility] conditions. {ISO 3534-1}
Repeatability conditions: Conditions where test results are obtained with the same method on identical test items in the same laboratory by the same operator using the same equipment within short intervals of time. {ISO 3534-1}
Reproducibility conditions: Conditions where test results are obtained with the same method on identical test items in different laboratories with different operators using different equipment. {ISO 3534-1}
Note:
When different methods give test results that do not differ significantly, or when different methods are permitted by the design of the experiment, as in a proficiency study or a material-certification study for the establishment of a consensus value of a reference material, the term “reproducibility” may be applied to the resulting parameters. The conditions must be explicitly stated.
Repeatability [Reproducibility] standard deviation: The standard deviation of test results obtained under repeatability [reproducibility] conditions. {ISO 3534-1}
Notes: {ISO 3534-1}
1. It is a measure of the dispersion of the distribution of test results under repeatability [reproducibility] conditions.
1. Similarly “repeatability [reproducibility] variance” and “repeatability [reproducibility] coefficient of variation” could be defined and used as measures of the dispersion of test results under repeatability [reproducibility] conditions.
Repeatability [Reproducibility] limit: The value less than or equal to which the absolute difference between two test results obtained under repeatability [reproducibility] conditions may be expected to be with a probability of 95%. {ISO 3534-1}
Notes:
1. The symbol used is r [R]. {ISO 3534-1}
1. When examining two single test results obtained under repeatability [reproducibility] conditions, the comparison should be made with the repeatability [reproducibility] limit
r [R] = 2.8 sr[sR]. {ISO 5725-6, 4.1.4}
3 When groups of measurements are used as the basis for the calculation of the repeatability [reproducibility] limits (now called the critical difference), more complicated formulae are required that are given in ISO 5725-6:1994, 4.2.1 and 4.2.2.
Estimation
The calculated repeatability and reproducibility values can be compared with existing methods and a comparison made. If these are satisfactory then the method can used as a validated method. If there is no method with which to compare the precision parameters then theoretical repeatability and reproducibility values can be calculated from the Horwitz equation for concentrations down to 120 µg/kg or the modified equation at levels less than 120 µg/kg and greater than 13.8%.
i.e.
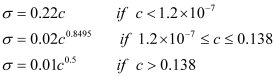
Proportion of the amount of analyte present or added to the test material which is extracted and presented for measurement.
Analytical methods do not always measure all of the analyte of interest present in the sample. Analytes may be present in a variety of forms in samples not all of interest to the analyst. The method may thus be deliberately designed to determine only a particular form of the analyte. However a failure to determine all of the analyte present may reflect an inherent problem in the method. Either way, it is necessary to assess the efficiency of the method in detecting all of the analyte present.
Because it is not usually known how much of a particular analyte is present in a test portion it is difficult to be certain how successful the method has been at extracting it from the matrix. One way to determine the efficiency of extraction is to spike test portions with the analyte at various concentrations, then extract the fortified test portions and measure the analyte concentration. The inherent problem with this is that analyte introduced in such a way will probably not be held as strongly as that which is naturally present in the test portion matrix and so the technique will give an unrealistically high impression of the extraction efficiency. It is however the most common way of determining recovery efficiency, and it is recognised as an acceptable way of doing so. However the drawback of the technique should be borne in mind. Alternatively it may be possible to carry out recovery studies on reference materials, if suitable materials are available. Provided these have been produced by characterisation of natural materials rather than by characterisation of synthetic materials into which the analyte has been spiked, then the recovery study should accurately represent the extraction of real test portions.
Recoveries - Quick Reference | |||
Analyse |
Repeats |
What to calculate from the data |
Comments |
Matrix blanks or samples unfortified and fortified with the analyte of interest at a range of concentrations |
6 |
Determine recovery of analyte at the various concentrations.
|
Fortified samples should be compared with the same sample unfortified to assess the net recovery of the fortification.
|
Certified reference materials (CRM) |
Determine recovery of analyte relative to the certified value |
Depending on how the CRM was produced and characterised, it may be possible to get >100% recovery. | |
SELECTIVITY
Definition
Selectivity is the extent to which a method can determine particular analyte(s) in mixtures or matrices without interferences from other components.
Selectivity is the recommended term in analytical chemistry to express the extent to which a particular method can determine analyte(s) in the presence of interferences from other components. Selectivity can be graded. The use of the term specificity for the same concept is to be discouraged as this often leads to confusion.
Selectivity/specificity are measures that assess the reliability of measurements in the presence of interferences. The selectivity of a method is usually investigated by studying its ability to measure the analyte of interest in test portions to which specific interferences have been deliberately introduced (those thought likely to be present in samples). Where it is unclear whether or not interferences are already present, the selectivity of the method can be investigated by studying its ability to measure the analyte compared to other independent methods/techniques.
Confirmation of identity and selectivity/specificity - Quick Reference | |||
What you do |
How many |
Calculate / determine |
Comments |
Analyse samples, and reference materials by candidate and other independent methods. |
1 |
Use the results from the confirmatory techniques to assess the ability of the method to confirm analyte identity and its ability to measure the analyte in isolation from other interferences. |
Decide how much supporting evidence is reasonably required to give sufficient reliability. |
Analyse samples containing various suspected interferences in the presence of the analytes of interest. |
1 |
Examine effect of interferences – does the presence of the interferent enhance or inhibit detection or quantification of the measurands. |
If detection or quantitation is inhibited by the interferences, further method development will be required. |
SENSITIVITY
Definition
Change in the response divided by the corresponding change in the concentration of a standard (calibration) curve; i.e., the slope, si, of the analytical calibration curve.
Note:
This term has been used for several other analytical applications, often referring to capability of detection, to the concentration giving 1% absorption in atomic absorption spectroscopy, and to ratio of found positives to known, true positives in immunological and microbiological tests. Such applications to analytical chemistry should be discouraged.
Notes: {IUPAC-1987}
1. A method is said to be sensitive if a small change in concentration, c, or quantity, q, causes a large change in the measure, x; that is, when the derivative dx/dc or dx/dq is large.
1. Although the signal si may vary with the magnitude of ci or qi, the slope, si, is usually constant over a reasonable range of concentrations. si may also be a function of the c or q of other analytes present in the sample.
Estimation
This is effectively the gradient of the response curve, i.e. the change in instrument response that corresponds to a change in analyte concentration. Where the response has been established as being linear with respect to concentration, i.e. within the linear range of the method, and the intercept of the response curve has been determined, sensitivity is a useful parameter to calculate and use in formulae for quantitation. Sensitivity is sometimes used to refer to limit of detection but this use is not generally recommended.
[Note: much of the detailed recommendations in Appendix VII have been taken from published texts, specifically:
AOAC-I Peer Verified Methods, Policies and procedures, 1993, AOAC International, 2200 Wilson Blvd., Suite 400, Arlington, Virginia 22201-3301, USA.
W. J. Youden; Steiner, E. H. ‘Statistical Manual of the AOAC-Association of Official Analytical Chemists’, AOAC-I, Washington DC, 1975, p35.
“The Fitness for Purpose of Analytical Methods: A Laboratory Guide to Method Validation and Related Topics” Eurachem Guide, 1998, http://www.eurachem.ul.pt/guides/valid.pdf.
Nomenclature in evaluation of analytical methods, including detection and quantification capabilities (IUPAC Recommendations 1995). Pure & Appl. Chem., 1995, 67, 1699-1723.
Detection in Analytical Chemistry – Importance, Theory and Practice. L. A. Curries, ACS Symposium Series 361, American Chemical Society, Washington DC 1988. Various chapters are recommended, particularly Ch4 (Kirchmer, C. J.) and Ch 16 (Kurtz, D. A. et al.)
Analytical Methods Committee, “Recommendation for the Definition, Estimation and Use of the Detection Limit”, The Analyst, 1987, 112, 199-204.
“Evaluation of Analytical Methods used for Regulation of Foods and Drugs”, W. Horwitz, Anal. Chem. 1982, 54 (1), 67A - 76A.
M. Thompson, Analyst, 2000, 125, 385-386.]
NMKL Procedure No. 9
