Enzyme-linked immunosorbent assay (ELISA)
S.M. Garnsey and M. Cambra
This guide to the use of enzyme-linked immunosorbent assay (ELISA) is based on the chapter on ELISA in the handbook Detection and diagnosis of graft-transmissible diseases of citrus, but with a few minor changes for specificity to grapevine. It describes several common variations of ELISA. Some background information is presented to help the user understand the technique and make modifications to this highly flexible procedure for specific applications. Information on the selection of techniques, on preparation of samples for testing and on the basic steps of the ELISA protocol is provided. The materials, reagents and equipment needed are indicated, and some specific examples are given.
Properly used, ELISA is a sensitive, accurate and rapid detection method. It is especially effective when large numbers of samples must be assayed, when results are needed rapidly and when suitable indicator plants and/or greenhouse facilities are not available. ELISA has been developed for grapevine fanleaf virus and a number of other mechanically and nonmechanically transmissible viruses such as grapevine virus A, leafroll-associated closterovirus I, II and III and fleck-associated isometric virus, as well as for flavescence dorée MLOs and Xvlella fastidiosa, the agent of Pierce's disease.
ELISA is simple and can be carried out by most people after brief training and some practice. As with any indexing procedure, some experience is necessary to use ELISA accurately and confidently. New users should consult several of the excellent general references on ELISA (e.g. Clark and Bar-Joseph, 1984; Clark, Lister and Bar-Joseph, 1988; Sanchez-Vizcaino and Cambra Alvarez, 1987) which provide additional details on theory and application. It is very useful to visit a laboratory where ELISA is practiced in order to observe the procedure and to practice it under the guidance of an experienced user. Begin with a well-known system, and study the effects of adjusting reactant concentrations and test conditions.
Extensive training and background in serology and immunology are not essential to use ELISA, but understanding of some basic concepts is necessary. ELISA is a serological technique, and like other serological procedures it is based on the concept that many proteins are antigenic when injected into animals and that the immunized animal will form antibodies to them.
These antibodies can be obtained from the serum of the immunized animal and will react specifically with the antigen to which they were formed. A primary requirement to begin ELISA is a useful source of antibody to the pathogen to be detected. This in turn means that antigen specific to the pathogen must be identified and purified sufficiently to produce the needed antibodies. Antigen purification and antibody production are beyond the scope of this section, but information on these topics is contained in some of the references cited (Clark, Lister and Bar-Joseph, 1988; Van Regenmortel, 1982).
Also fundamental to ELISA is the concept that various enzymes can be bound to antibody molecules to form a conjugated molecule that has enzymatic activity and is also serologically active. Since enzymes are highly active and can be detected at low concentrations, they are effective labels. Enzyme-labelled antibodies can be detected when they are exposed to a substrate that enzymes can change. Normally, a substrate that changes colour as a result of the enzyme action is used. The amount or rate of colour change can then be used to measure the amount of antibody present. Enzyme labels provide a sensitivity similar to that of radioactive labels and have several important advantages: they are stable, inexpensive and safe to use, and they can be used successfully without sophisticated equipment.
The enzyme label may be attached directly to the antibody used to detect the antigen in question (the detecting antibody). This is called a direct assay, of which the highly popular double antibody sandwich technique described below and illustrated in Figure 262a is a good example. The label may also be used indirectly. In this case, the label is attached not to the detecting antibody, but rather to a second antibody specific to the detecting antibody. Antibodies of one species are antigenic when injected into an animal of a second species. For example, rabbit immunoglobulins can be injected into another animal such as a goat to create a goat anti-rabbit antiserum. These goat anti-rabbit antibodies are useful to detect antibodies from rabbits that were originally prepared to detect another antigen.
Indirect assays are more sensitive and also avoid the need to prepare a conjugate to each antibody used. Several forms of indirect assay are described in the following section and are also illustrated in Figure 262 (b and d). The relative advantages of direct and indirect systems are discussed in the following section.
Several other molecular interactions are frequently used in conjunction with ELISA, either to purify immunoglobulins or to amplify reactions and increase sensitivity. Protein A is a cell wall component of the bacterium Staphylococcus aureus and has the unique characteristic of binding to the immunoglobulin protein of many mammalian species. The binding site is on the Fc region of the immunoglobulin and not on the antigen binding site. Protein A is frequently used to purify antibodies by affinity chromatography. It can also be conjugated with enzymes and used in assays to detect immunoglobulins.
A second important system is the biotin/avidin system. Biotin, a small vitamin, has a very high affinity for avidin, a 68 000 molecular weight glycoprotein. Antibodies and enzymes can be conjugated with several molecules of biotin to form a "biotinylated" molecule. Each avidin molecule has four binding sites for biotin. This multiplying interaction has been exploited in several ways to amplify the number of enzyme molecules associated with each antigen-bound antibody and thereby increase sensitivity. One example is illustrated in Figure 262c.
Another fundamental concept for ELISA is that proteins such as antibodies and virus coat proteins will adsorb strongly to the surface of certain plastics such as polystyrene and polyvinyl chlorides. Protein binding also occurs to some forms of cellulose nitrate. These materials are frequently referred to as "immunosorbents" or the "solid phase" in ELISA protocols. The protein binding to immunosorbent materials is not specific and is not a serological reaction such as occurs between antigen and antibody molecules. If a mixture of antibodies is exposed to an immunosorbent plastic, all will bind. Similarly, when a crude extract from a diseased plant is placed in an ELISA plate, proteins of the pathogen and proteins of the host present in the extract will both be bound.
Binding either the antibody or the antigen component of a serological system to a solid phase is very useful because the bound component can subsequently be used to probe complex mixtures of potential reactants. Only those that are serologically related will be trapped. All nonreactive components can then be removed by washing and will not interfere with subsequent steps. For example, when an extract from a virus-infected plant is placed in the wells of a microtitre plate coated with antibodies to that virus, virus antigens in the extract will be bound to the trapping antibody and all nonrelated proteins will be removed by the subsequent washing step.
Undesired adsorption of antibody or antigen proteins to the plastic can be avoided by using non-ionic detergents such as Tween 20 in incubating solutions or by adding an excess of a non-specific protein to block all sites not occupied by the desired serological component. For example, the buffer used to coat plates with trapping antibody does not contain Tween 20, but Tween 20 is incorporated in subsequent steps where any non-specific binding of other proteins should be avoided.
Immunoblotting procedures are not specifically discussed in this section. However, much of the information and the general concepts presented are directly applicable to immunoblotting procedures. The main differences are that the solid phase for immunoblotting is usually cellulose nitrate, the substrate used to measure presence of the antigen-antibody-enzyme complex is different and incubation conditions may be somewhat modified.
ELISA PROCEDURES
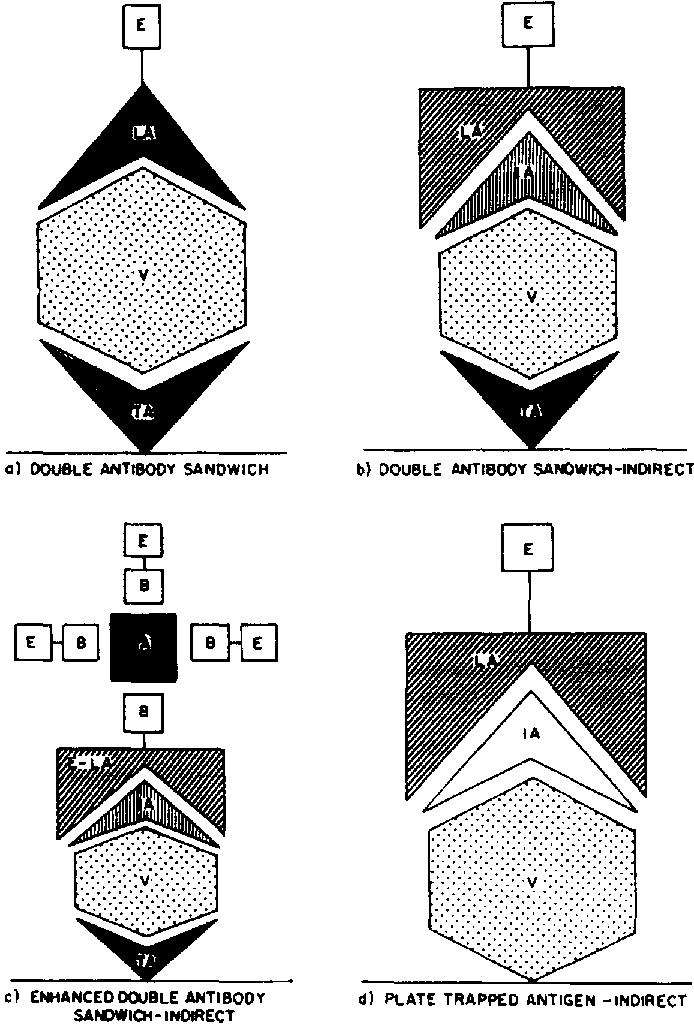
Numerous variations of the ELISA procedure can be devised (Clark, Lister and Bar-Joseph, 1988; Engvall and Pesce, 1978; Jones and Torrance, 1986; Koenigand Paul, 1982; Maggio, 1980). The selection depends on the sensitivity, specificity and convenience required; the presence of interfering factors; and the type and activities of the antisera available. The basic steps for four commonly used variations of ELISA are outlined here and illustrated in Figure 262. In three of the variations, a, b and c, the solid-phase (ELISA plate) is coated with antibody to the antigen to be detected. This antibody, identified as trapping antibody (TA in the figure), then traps its corresponding antigen (identified as V) from suspension or solution. In the fourth variation (Figure 262d), the antigen (V) is trapped directly on the solid phase and detected with its specific antibody.
Double antibody sandwich
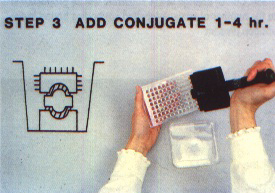
The inexperienced user should start with the double antibody sandwich (DAS) where possible. This has been the most commonly used form of ELISA for plant virus detection since its description by Clark and Adams (1977). The components of DAS are illustrated in Figure 262a. The immunosorbent surface is a plastic microtitre plate with wells designed for ELISA as shown in Figure 263. A dilute solution of unlabelled antibody is added to the wells of the plate (Figure 264), and the antibody adsorbed on the plastic becomes the trapping antibody (TA) as illustrated in Figure 262a. After washing to remove any excess antibody (see Figure 277), the sample (antigen) is added as shown in Figure 265. Antigens (V in Figure 262) specific to the bound trapping antibody attach themselves to it, but other proteins remain in solution and are removed by washing. The antigen attached to the trapping antibody is detected by adding a labelled antibody (LA in Figure 262a) specific to the antigen (Figure 266). The label is the enzyme (E) previously conjugated to the antibody. When substrate specific to the enzyme is added in the final step (Figure 267), a colour develops as a result of enzyme action (Figures 268 to 270). The amount of colour and its rate of development are correlated to the amount of labelled antibody bound to the antigen which had been trapped by the antibody attached to the plate.
DAS can be done with a single good quality polyclonal antiserum. The immunoglobulins present are partially purified, and one portion is saved for use as trapping antibody while another is conjugated to an enzyme. Alkaline phosphatase is commonly used as the enzyme and the conjugation can be done in the presence of dilute glutaraldehyde (Clark, Lister and Bar-Joseph, 1988). The antibodies for coating and detection do not have to come from the same source, e.g. monoclonal antibodies could be used for coating and a polyclonal antiserum could be used to prepare the enzyme-labelled antibody.
Double antibody sandwich indirect
DAS can be converted to an indirect procedure (DAS-I), which is illustrated in Figure 262b. The first two steps are the same as in DAS. However, the antigen bound to the trapping antibody is detected by an unlabelled intermediate antibody (IA) which is specific to the same antigen but is from an animal species different from the one used to prepare the trapping antibody. For example, if the trapping antibody was prepared in rabbits, the detecting or intermediate antibody could be from a mouse or a chicken. The unlabelled IA which attaches to the antigen is detected by an enzyme-labelled antibody (LA) specific to the IA. Because the IA is from a different species than the TA, the LA binds only to the IA and no non-specific binding of the LA to the TA occurs. The amount of LA is measured by adding substrate and measuring colour change as in DAS.
DAS-I ELISA involves an additional step (Figure 262b) but is more sensitive and also allows use of a commercially prepared enzymelabelled antibody to the IA. A single LA can also be used for multiple virus detection systems. In addition, the intermediate antibody does not have to be purified and is needed in only a limited quantity. If the intermediate antibody is highly specific, e.g. most monoclonals, then a highly specific antiserum is not required for coating. The major problem is that antibodies to the same antigen must be prepared in two different animals. If the trapping and the intermediate antibodies are from the same species, the labelled antibody used to detect the intermediate antibody will also bind to the trapping antibody and result in a nonspecific response.
A system has been devised to carry out DAS-I using a single antiserum (Adams and Barbara, 1982; Clark, Lister and Bar-Joseph, 1988). To do this, the antibodies are treated with the enzyme pepsin to remove the Fe portion of the molecule. The remaining F(ab'), fragment still has the antigen binding sites and will bind to the immunosorbent, but will not bind to protein A. The F(ab'), fragments are used as trapping "antibody" and the whole antibody is used as the intermediate antibody. Enzyme-conjugated protein A is then used instead of a labelled antibody to detect the intermediate antibody. It does not react to the trapping "antibody" because the Fe region has been removed.
The DAS-I procedure can be further modified to amplify the reaction achieved. This is commonly done using a biotin-avidin interaction in which the labelled antibody is biotinylated to react with avidin molecules conjugated to multiple enzyme molecules, as illustrated in Figure 262c. Different types of amplification are possible and special kits may be purchased to perform them. Users should be aware of the possibility of employing amplification when additional sensitivity is needed, but regular procedures should be tested before amplification is attempted.
Plate-trapped antigen
Another basic approach to ELISA is the platetrapped antigen procedure (Figure 262d). The approach is to trap the antigen (V) on the plastic surface, then react the trapped antigen with an unlabelled intermediate antibody (IA) specific to it. The IA is then detected as in DAS-I using an enzyme-labelled antibody (LA) specific to the IA. This procedure, called plate-trapped antigen indirect (PTA-I) ELISA, is relatively simple and involves no advance purification of antisera or conjugate preparation if a commercially prepared enzyme-labelled antibody to the unlabelled IA is used. The PTA-I procedure is usually less sensitive than DAS or DAS-I for use with crude plant extracts and may not be effective when antigen concentration in the sample is low. Since binding to the plate is non-specific, the target antigen and other proteins present in the extract compete for the available binding sites on the plate. Platetrapped antigen tests can be conducted as a direct assay using an enzyme-labelled antibody to the antigen, but sensitivity is even lower than for the indirect method, and the conjugate must still be prepared. Amplification procedures as described for DAS-I can also be used for the PTA-I procedure to increase sensitivity.
The specific steps and schedules for these types of ELISA are described in Schedules 1 to 3.
SAMPLING
Selection of appropriate samples for testing is critical. Although ELISA is a sensitive procedure, reliable results may not be obtained if poor samples are tested. Virus titre in grapevine tissue often varies markedly, and thousandfold differences in antigen concentration can occur over a relatively short period. Virus concentrations are generally highest in young, expanding flush tissues. They decrease rapidly as tissues mature under hot-weather conditions and more slowly under cool conditions. Avoid sampling old, mature tissue during the summer months in hot climates unless preliminary testing indicates that reliable samples can be taken. If the virus or pathogen is phloem-limited (e.g. the grapevine pathogens leafroll, rugose woodassociated closteroviruses and fleckassociated isometric virus), then the tissue sample collected must contain phloem tissue. Older bark tissue can be sampled if the cambium is active, but generally it is less reliable than young shoot flush, bark or young leaf midribs (Figures 271 and 272). Young root tips may be useful under some conditions.
A composite sample from several sites on the vine should be collected; normally three to five locations per vine are sampled. Increase sampling if the pathogen is irregularly distributed or when trying to monitor a recent infection.
Fresh tissue can normally be stored for at least seven to ten days at 4°C when kept in a plastic bag or sealed container.
Samples can also be stored frozen at -20°C or below for extended periods. Unprocessed fresh tissue should be used; or tissue can be diced, placed in extraction buffer and frozen in the grinding tube (Figure 273). The sample should not be ground prior to freezing because fresh extracts often lose much activity when frozen. Frozen samples should not be stored in an automatically defrosting freezer. Extracts can be stored for long periods when freeze-dried; this is a good way to store a source of consistent reference (control) samples. Always test a storage method with the specific pathogen under study in order to prove its effectiveness.
EXTRACTION
Numerous buffers and different additives have been used for extraction of tissue samples with different virus-host systems (Bar-Joseph and Garnsey,1981; Clark, 1981; Clark and Bar-Joseph, 1984; Clark, Lister and Bar-Joseph, 1988; McLaughlin et al., 1981). Phosphatebuffered saline (PBS) or 0.05 M Tris, pH 7.5 to 8.0, without any additives usually give good results for sandwich assays. Additives such as polyvinylpyrrolidone (PVP), EDTA and DIECA are generally unnecessary for citrus, but with grapevine tissues 2.5 percent nicotine or 2 percent PVP may be useful (see chapter on grapevine degeneration - fanleaf). Test the effect of additives before using them routinely. For plate-trapped antigen procedures, try extraction of the sample in carbonate coating buffer, pH 9.6, or in 0.05 M Tris, pH 8.0. Do nor use Tween 20 in the extraction buffer for samples to be plate-trapped.
Normally, the ratio of buffer to sample tissue should be at least 1: 10. Higher concentrations of tissue may actually reduce reaction rates and make sample preparation more difficult.
There are many ways to grind samples. Pestle and mortar are fine for small numbers of tender samples. Addition of an abrasive, such as fine sand or carborundum, to the sample or powdering the tissue in liquid nitrogen makes grinding easier. A dispersion homogenizer (Figure 274) equipped with a 10 to 25 mm diameter shaft is a good choice when large numbers of samples are to be processed. A 2 to 10 ml sample can be rapidly ground in a testtube or centrifuge tube of suitable diameter and length with this type of homogenizer. Fibrous tissue, such as bark and leaf midribs, should be cut into short pieces (2 to 5 mm) prior to grinding or the shaft will become clogged with fibre and need to be cleaned between samples. Two rinses of the grinder shaft in 500 to 1 000 ml clean water are usually adequate (Figure 275). Run the homogenizer briefly in each rinse solution.
Chill samples prior to grinding to offset heating during the grinding process. It is normally not necessary to keep the sample on ice during grinding, unless unusually long grinding is required and the sample becomes warm to the touch. Frequent users of dispersion homogenizers should wear earplugs to protect their hearing, and homogenizers should be isolated.
If necessary, samples can be prepared with very minimal equipment. When virus concentration is high, extensive disruption of the sample tissue is usually unnecessary. One method is simply to place a small piece of tender tissue directly in buffer in the well of an ELISA plate with forceps and then squeeze it to release the cell contents. Tissue can also be crushed in a small plastic bag using a mallet or smooth stone and the extract moved by pipette into the test plate.
Samples containing a lot of debris after extraction can be difficult to pipette. Remedies include centrifugation of the sample to pellet the debris or filtering the sample through a coarse filter such as cheesecloth or glass wool (Figure 276). Cutting off a portion of the tapered tip of a plastic pipette creates a wider orifice and is often quick and effective. It is frequently quicker to rinse a repeating pipette between samples than to change tips, so only a limited number of tips need be modified.
WASHING
Proper washing of the plates between steps is important. The standard procedure is to wash the plate three times between each step with phosphate-buffered saline (PBS) containing 0.5 percent Tween 20 (PBST). Sodium azide is frequently included in PBST solutions as a preservative. However, it is highly poisonous and may form an explosive complex with some metals. Sodium azide is unnecessary in ELISA wash solutions and should be omitted. The two most critical wash steps are after sample incubation when cross-contamination must be avoided between wells containing different samples (omit the first wash immediately if carry-over between wells occurs) and after the conjugate incubation step. If even a minor residue of unattached conjugate remains, high background readings may occur. (Add another wash at this point when in doubt.) Various plate washers are available which can promote consistent washing operations, but a plastic squeeze bottle will work well for small volumes of plates (Figure 277). Solutions in the plate wells can be removed by aspiration to avoid contamination, but usually the plate is inverted rapidly with a quick shake of the hand and tapped firmly on clean blotting paper or paper towels.
TEST CONDITIONS
A wide variety of reactant concentrations and incubation times and conditions have been reported for ELISA (Clark and Bar-Joseph, 1984; Clark, Lister and Bar-loseph, 1988; McLaughlin et al., 1981). The choice of conditions depends to some extent on the basic goals. With high concentrations of reactants, short incubation times can be used, and if necessary the entire ELISA procedure can be completed within two hours. Increasing the incubation time while decreasing concentration (especially of the conjugate) will conserve reactants. Reactions occur most rapidly at 30 to 37°C, but room temperature (20 to 28°C) will give satisfactory results. Gentle shaking during incubation may improve efficiency. Many workers find it convenient to do the sample incubation step overnight and often do this at 4 to 6°C.
Some experimentation will be necessary to determine optimum conditions for each situation. Schedules 1 to 3 give examples which should provide a good starting point. Moderate changes in times and conditions are unlikely to cause a test failure, and changes can often be made to render the schedule more convenient for the user with no loss of information. New users should certainly experiment with different schedules to find the optimum for their purpose.
One of the major variables in ELISA to be evaluated is the concentration of conjugate to use. Commercially prepared enzyme-labelled antibodies normally have a recommended working dilution (frequently between 1/1 000 and 1/2 000). Conjugates that are prepared experimentally may differ markedly, and published values for other systems are of little help. Optimum dilutions of 1/100 to 1/20 000 of the stock preparation (approximately 1 mg per ml) have been reported. The effective dilution will depend on the basic affinity of the antibody, the titre of specific and non-specific (host) antibodies, the source and activity of the enzyme used and the effectiveness of the conjugation procedure. When starting with a new or unknown batch of conjugate, test three tenfold dilutions starting at a 1/100 dilution to determine the approximate activity. Using these results, make a second test in the appropriate dilution range indicated. Normally, the objective is to obtain a strong positive reaction to a good positive sample within 20 to 60 minutes and little or no reaction to healthy extracts. If the conjugate concentration is so high that a reaction is instantly visible, a background reaction is often also observed with healthy extracts (and even with buffer controls). Reduce conjugate concentration and, if a nonspecific reaction persists, adsorb the antiserum against a concentrated extract of healthy plant tissue to remove antibodies against healthy antigens. If possible, do this before purifying the IgG. Adding healthy tissue extract to the buffer used to dilute the conjugate may also reduce nonspecific reactions (Clark, Lister and Bar-Joseph, 1988).
The use of appropriate controls is essential. Each plate should have at least one healthy and one known positive sample as controls. A buffer control is also useful to determine the level of background reaction to healthy extracts. Frequently, a slightly higher reading will be observed for the buffer control than for the healthy extract because proteins in the extract block exposed protein binding sites on the plastic, which may later non-specifically bind conjugate molecules. Each sample should be tested in at least two wells. A random loading pattern can be used, but paired wells are normally used for routine work. Special applications may require additional replication and randomization.
Edge effects in the plates were frequently noted when ELISA first became popular and outer wells were avoided. Plates have steadily improved and normally all wells can be used. Uniformity in new lots of plates can be checked by loading a uniform sample in all wells.
RECORDS
One of the major tasks of any indexing procedure is to identify samples properly during the testing process and to record results in a usable format.
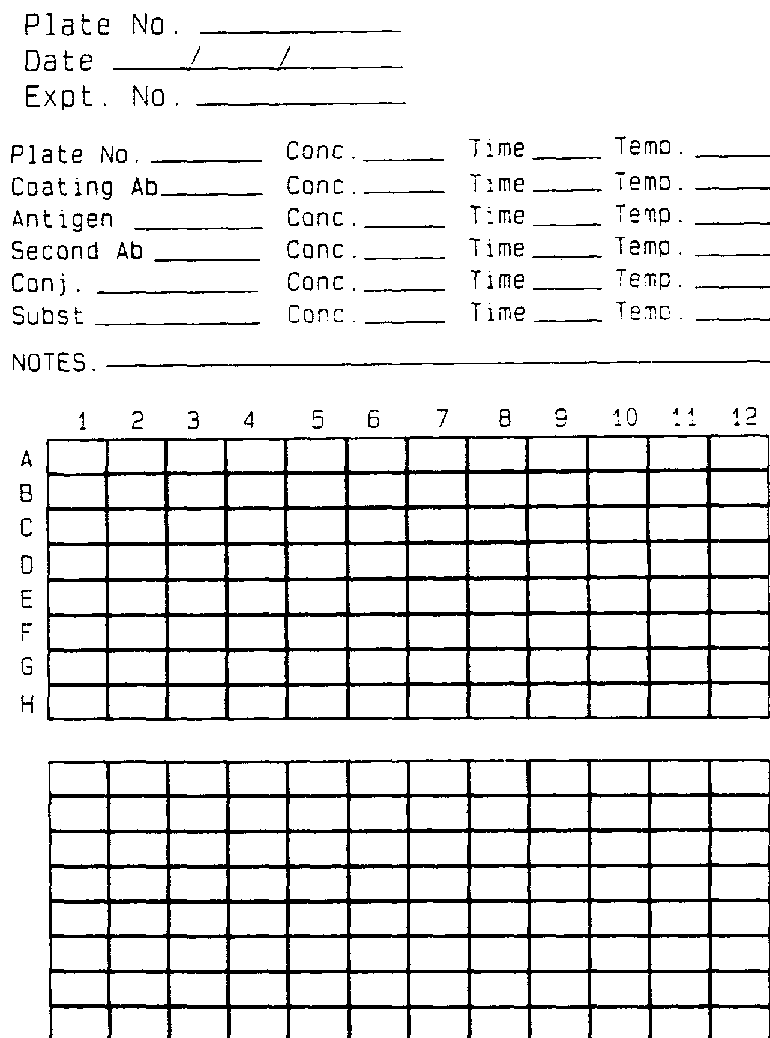
The identity of the sample must be maintained through the multiple steps of collection, processing, extraction and testing. It is usually convenient to give each sample a code number at the time of collection and to use this code during the test process. If samples are collected directly in the grinding vessel (usually a glass or plastic tube), labelling steps can be reduced. If the sample is collected in a container other than the grinding vessel, a transferable label is often convenient. Many ELISA plates have a coding system on the plate margins to identify individual wells, but there is no space to mark individual wells on the plate. Most workers develop a data sheet similar to the one shown as Figure 279, which is used to record the loading sequence of test samples and other pertinent data such as reactant concentration and incubation conditions. Each plate and data sheet should have a corresponding number recorded in a logbook to facilitate retrieval of information.
It is important to mark the loading pattern for each plate prior to loading samples and to arrange the samples to be loaded in the appropriate sequence. Note changes or errors that may occur during loading, and store tubes and samples under refrigeration until the testing process is complete.
Visual readings of the plate can be recorded directly on the plate data sheet. Printouts from a plate reader (Figure 270) can also be attached to the original data form. Use of computers to store and analyse data is increasing rapidly. Computers are convenient for long-term storage of large amounts of data. Data from the reader may also be converted into another format for further analysis and spreadsheet presentation on the computer. Permanent visual records of important tests can also be obtained by photographing individual plates on a light box or over a white background. A 35-mm transparency film is economical, and a standard exposure can be obtained if a constant light source is used.
EVALUATION OF RESULTS
Evaluation of ELISA results often presents some problems to the novice user. With highly specific antisera and samples with good antigen titre, results are normally very clear. When the antisera used are weak or contain some antibodies to host proteins and/or the samples have a very low antigen titre, determination of a positive result can be more difficult. Reactions can be evaluated visually with some precision if background readings for healthy controls are low. Normally, the eye can discern differences in OD405 of 0.05 to 0.1 above a low background. A graded scale with three to five levels is useful to report the relative degree of reaction. Where greater accuracy is required, the degree of reaction can be measured by testing a diluted sample in a spectrophotometer or by reading the plate in an ELISA plate reader (Figure 270). A wide variety of plate readers are available, from simple manual models suitable for modest numbers of plates to highly automated models capable of various levels of data analysis and storage.
Recently, emphasis has increased on measuring reaction rate rather than a single final optical density value. This eliminates some sources of error where accurate quantitative data are needed and also allows more accurate comparison of samples with large differences in antigen concentration. Rate calculation requires several measurements of the same plate at a measured time interval, and a plate reader is essential. Some plate readers do rate calculations automatically. Expensive plate readers are not necessary until a definite need for them is identified.
The limits of reliable detection are correlated to the precision used and the number of replications. Confidence levels can be calculated statistically when in doubt. Most workers establish an arbitrary threshold value relative to the healthy control for a positive reaction, such as a reading twice that of the healthy control or the healthy reading plus 0.1 OD. Where reaction to healthy extracts is low (<0.05 OD), the eye can usually consistently detect reactions of 0.1 OD or higher and this becomes the effective limit for visual recording. It is best to establish a conservative threshold rating and retest all questionable samples. Some experimentation with different dilutions of known samples will help.
PURIFICATION OF IMMUNOGLOBULINS
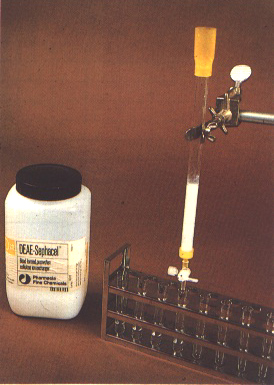
Numerous procedures are now available for purification of immunoglobulins (IgG) from polyclonal antisera or for purification of monoclonal antibodies from culture fluid or ascites fluid. The easiest method is to use a commercial kit or system containing detailed instructions. Many of the kits are based on separation of the IgG component by protein A bound to a solid substrate. The IgG is subsequently eluted from the protein A by changing buffers and collected. An alternative, which is slower but inexpensive, is to precipitate the IgG from solution by use of ammonium sulphate and then fractionate the dialysed resuspended pellet by column chromatography on a DEAE cellulose column (Figure 278).
Purified IgG solutions are normally adjusted to a concentration of 1 mg per ml (equal to an OD280 of 1.4 when measured spectrophotometrically) and stored at 4°C in PBS containing at least 0.02 percent sodium azide. IgG preparations can also be stored in 50 percent glycerol at -20°C or freeze-dried. More detailed instructions can be found in Clark, Lister and Bar-Joseph (1988).
PREPARATION OF ENZYME-LABELLED ANTIBODIES
Conjugated molecules of antibody and enzyme can be prepared in several ways (Bar-Joseph and Garnsey, 1981; Clark and Adams, 1977; Clark and Bar-Joseph, 1984; Clark, Lister and Bar-Joseph, 1988; Engvall and Pesce, 1978). Alkaline phosphatase is the most widely used enzyme, and the single-step glutaraldehyde method is commonly used to prepare alkaline phosphatase conjugates. Test this system first before experimenting with other procedures. Alkaline phosphatase is usually obtained commercially as an enzyme precipitate in a salt solution. The precipitate is recovered from solution by centrifugation and about 5 mg is dissolved in 2 ml of a 1 mg per ml solution of IgG. The mixture is dialysed thoroughly to remove excess salts and then fresh glutaraldehyde is added to a final concentration of 0.05 percent. After 4-hour incubation, the conjugate is dialysed three times to remove the glutaraldehyde and stored at 4°C in PBS containing 0.04 percent sodium azide and 5 mg per ml bovine serum albumen. Use an enzyme source with a quality suitable for ELISA. Careful dialysis is very important for good results. New lots of conjugate must be tested to determine optimum dilutions for use (Sanchez-Vizcaino and Cambra Alvarez, 1987). Good-quality conjugates can normally be used at at least 1/500 dilution and dilutions as great as 1/5 000 or more may be possible. See Clark, Lister and Bar-Joseph ( 1988) for further details and for preparation of horseradish peroxidase conjugates.
Conjugates are stable for long periods at 4°C. Freezing or freeze-drying are not recommended unless preliminary testing indicates it is possible. If freezing is necessary, add 50 percent glycerol.
BASIC SUPPLIES AND EQUIPMENT
Sources of supplies and equipment change rapidly and new equipment continues to appear. It is not possible to list all suppliers in every country. To be of help, we have indicated some possible sources for some essential supplies. It is not essential to use these specific sources, and the user may, in fact, find a more convenient and economical local source than those listed. The Laboratory buyer's guide, available from International Scientific Communications, Inc., PO Box 870, Shelton, CT 06484, USA, is a useful directory of manufacturers. Basic equipment and supplies are emphasized rather than some of the more sophisticated and expensive equipment also available for largescale clinical work. It is assumed that most users of the latter already know the sources of supply.
A source of antiserum to the pathogen you wish to detect is essential. Both polyclonal and monoclonal antisera have been produced to a number of grapevine pathogens, and more are being developed. Polyclonal antisera are frequently preferable for general detection work where discrimination of a particular isolate is either unnecessary or undesirable. Unless the antigen used to produce the antiserum was well purified, polyclonal antisera frequently contain some antibodies to plant proteins as well as to the specific pathogen. It is advisable to check the specificity of the antiserum to be used in the initial stages to ensure acceptability. Monoclonal antibodies are generally more specific because, if properly prepared, only a single epitope of the antigen is involved. To do DAS-I assays, antibodies are needed from two animal species unless the F(ab')2 procedure is followed.
Production of high-quality antisera is frequently a time-consuming and difficult task, especially for people without experience in this process. Inexperienced users of ELISA should ordinarily try to obtain a small amount of antisera from existing sources for their initial work. Frequently, a modest supply of antiserum can be obtained from a colleague working on grapevine pathogens. Most grapevine virologists are members of the International Council for the Study of Viruses and Virus Diseases of the Grapevine (ICVG) and can be identified by writing to the Secretary, c/o Federal Agricultural Research Station of Changins, 1260 Nyon, Switzerland. Small amounts of purified IgG or enzyme-labelledconjugate may also be available for limited experimental tests.
Increasing access to antisera from commercial sources and the American Type Culture Collection can be expected. Current commercial outlets of ELISA supplies for grapevine pathogens include: Agdia Inc., 30380 County Road 6, Elkhart, IN 46514, USA; Ingenasa, Hermanos Garcia, Noblejas 41,28037 Madrid, Spain; Sanofi Sante Animale, Z.l. de la Ballastière, BP 126, 33500 Libourne, France; Bioreba AG, Gompenstrasse 8, 4008 Basel, Switzerland; Boehringer, Box 310120,6800 Mannheim, Germany; and Loewe Biochemia GmbH, Residenza Betulle 801,20090 Segrate (Ml), Italy.
If a prepared ELISA kit or purified sources of IgG and labelled conjugates are available, ELISA tests can be done in a very simple laboratory equipped with a balance, a simple pH meter, basic glassware, a refrigerator and a supply of deionized water. The other essential items are ELISA plates, several repeating pipettes and the chemicals to prepare the necessary buffers (see below). To purify IgG and to prepare antibodyenzyme conjugates, it is necessary to have access to a low-speed centrifuge, a UV spectrophotometer and some column chromatography equipment.
ELISA plates are available from numerous suppliers including Dynatech Laboratories, 14340 Sullyfield Circle, Chantilly, VA 22021, USA and Nunc Inc., 2000 N. Aurora Road, Naperville, IL 60540, USA. Plate quality can vary. Plates that work well for sandwich assays may work less well for plate-trapped antigen assays. If possible, find a reputable local supplier and test several types of plates before buying a large supply.
Several repeating pipettes, as shown in Figures 264 and 265, are more or less essential. Fixed volume models are economical and will suffice, but adjustable models are much more convenient and can be used for many other tasks. The minimum requirement is one pipette that will measure accurately in the 1 to 20,ul range (to make dilutions of IgG and conjugates) and one that will operate in the 100 to 1 000 ?l range (or 200 to 1000 ?l). Pipettes for the ranges of 20 to 200 ?l and 1 to 5 ml are also extremely useful. Multichannel pipettes (Figures 264 and 266) that can simultaneously dispense the same volume into 4 to 12 wells are very useful when many plates must be loaded. All repeating pipettes use disposable plastic tips. If possible, select pipettes that can use interchangeable tips. There are numerous manufacturers of pipettes and numerous models. Consult a laboratory supply company or manufacturers such as Flow Laboratories S .A., Lugano, Switzerland or Rainin Instruments Co., Woburn, MA 01801, USA for current information.
Plates can be read visually, but if large numbers of plates are to be done on a regular basis a plate reader greatly speeds reading and makes evaluation of results easier. Numerous models with varying degrees of automation are available and details change rapidly. It is not necessary, and probably not advisable, to buy a reader until the user has some initial experience and knows the system will work. Before purchase, ask for a demonstration and also consult users in other laboratories for their recommendations. Large-scale users should consider readers that are computer compatible.
See Part IV for additional information on laboratory equipment.
ELISA BUFFERS AND SOLUTIONS
A limited number of chemicals are required to make the buffers and solutions needed for ELISA, and these are shown in Table 7. Many of these should be readily available in most biological laboratories. Specific sources and catalogue numbers have not been listed, but suggestions can be obtained from other ELISA users. Sigma Chemical Co., St Louis, MO 6317X, USA; Boehringer Mannheim Biochemicals, Indianapolis, IN 46250, USA; and Pharmacia LKB Biotechnology AB, Uppsala, Sweden or Piscataway, NJ 08854, USA are useful general sources for chemicals, enzymes and antibodies mentioned in this section if satisfactory local suppliers cannot be located. See also the Laboratory buyer's guide mentioned above.
TABLE 7 List of chemicals for ELISA
| 1. Alkaline phosphatase type VlI | |
| 2. Bovine serum albumen | BSA |
| 3. DEAE cellulose | |
| 4. Diethanolamine | NH (CH2CH2OH)2 |
| 5. Glutaraldehyde | OCH(CH2)3CHO |
| 6. Hydrochloric acid | HCI |
| 7. Ovalbumen | |
| 8. 4-Nitrophenyl phosphate | |
| 9. Polyvinyl pyrrolidone MW 40 000 | |
| 10. Potassium chloride | KCI |
| 11. Potassium phosphate | KH2PO4 |
| 12. Sodium azide | NaN3 |
| 13. Sodium bicarbonate | NaHCO3 |
| 14. Sodium carbonate | Na2CO3 |
| ,15. Sodium chloride | NaCI |
| 16. Sodium hydroxide | NaOH |
| 17. Sodium phosphate (dibasic) | Na2HPO4 |
| 18. Tris(hydroxymethyl)aminomethane HCI | Tris-HCI |
| 19. Tween 20 |
The materials listed are for the alkaline phosphatase system. If horseradish peroxidase or another enzyme is used, make appropriate changes in substrate and substrate buffer.
The formulae for buffers and substrate solutions needed are shown in Table 8. Use glassdistilled or high-quality deionized water to prepare buffers and solutions. The formulae given in Table 8 are for one-litre quantities. Larger quantities of the wash buffer (PBST) than of other solutions are used. The dry salts needed for PBS can be weighed in advance in units to make a convenient volume, mixed dry and stored in sealed plastic bags until needed. A new supply of PBS can be obtained rapidly, as needed, by adding the required volume of distilled wafer to the weighed salts.
Use standard buffer solutions with pH values near 7.0 and 10.0 to calibrate pH meters. Store buffers (except PBST) at 4°C if possible.
SCHEDULES
Schedules are shown here for types of ELISA illustrated in Figure 262, a, b and d, to provide some specific examples of reactant concentration and incubation time and conditions. The details shown are those typically used with an alkaline phosphatase enzyme-label system. No schedule is shown for the amplified form of DAS-I illustrated in Figure 262c. The preliminary steps are the same as used for DASI and the schedule for the enhancement steps will vary with the enhancement protocol used. Specific instructions are generally provided with the enhancement materials when these are purchased in kit form.
TABLE 8 ELISA buffers and solutions
1. Coating butter
1.59 g Na2CO3
2.93 g NaHCO3
0.2 g NaN3
(pH should be 9.6)1
2. Phosphate buffered saline (PBS)
8 g NaCI
0.2 g KH2PO4
2.9 g Na2HPO4 12 H2O
(1.15 g anhydrous)
0.2 g KCI
(pH should be 7.2 to 7.4)'
3. Washing buffer (PEIST)
1,0 litre PBS
0.5 ml Tween 20
4. Extraction buffer
1.0 litre PBST
20 9 polyvinyl pyrrolidone, MW 40 0002
(Option: 15.7 9 Tris-HCI, adjusted to pH 7.8 with NaOH)
5. Conjugate buffer
1.0 litre PBST
20 g polyvinyl pyrrolidone, MW 40 0002
2.0 g ovalbumen
0.2 g NaN3
6. Substrate buffer
97 ml diethanolamine
0.2 g NaN3
(adjust pH to 9.8 by adding HCI)
7. Reaction stopping solution
120 g NaOH
1pH should be close to value, adjust slightly if
necessary.
2Poly vinyl pyrrolidone is not essential for
extraction or conjugate buffers.
Normally, 200 ml of solution are added per well, but smaller volumes can be used. If NaOH is used to stop the reaction, 50 ml are added to the wells already containing substrate.
Schedule 1
Double antibody sandwich ELISA
1. Coat ELISA plates (Figure 264) with antibodies (IgG) diluted to 1 to 2 ?g per ml in carbonate coating buffer. Incubate for 1 to 4 hours at 25 to 30°C and wash three times with PBST (Figure 277).
2. Add sample extracts (Figure 265) prepared at a 1/10 to 1/20 dilution in extraction buffer in duplicate or triplicate wells. Incubate for 2 to 4 hours at 30 to 37°C or overnight at 4 to 6°C. Wash thoroughly three times with PBST. Avoid cross-contamination of samples when washing.
3. Add enzyme-antibody conjugate diluted in conjugate buffer to an optimum concentration (normally between 1/500 and 1/5 000) (Figure 266). Incubate 2 to 4 hours at 37°C. Wash at least three times with PBST to remove unbound conjugate from the wells.
4. Add substrate freshly prepared at a concentration of 0.6 to 1 mg per ml in substrate buffer (10 percent diethanolamine, pH 9.8) (Figure 267). Incubate until strong colour change develops in positive controls (normally 30 to 60 minutes) (Figure 268) and read plates (Figure 270). Plates may be read at several intervals without stopping the reaction, to calculate reaction rate, or the reaction can be stopped at an appropriate time by addition of 3 M NaOH and a single reading can be made. If plates are read visually, score the estimated relative strength of reaction. If read on a spectrophotometer or with a plate reader (Figure 270), record the OD405 values.
Schedule 2
Double antibody sandwich indirect ELISA
1. Coat ELISA plates (Figure 264) with antibodies (IgG) specific to the antigen to be tested. The IgG concentration should be 1 to 2 mg per ml in carbonate coating buffer. Incubate for 1 to 4 hours at 25 to 30°C and wash three times with PBST (Figure 277).
2. Add sample extracts (Figure 265) prepared at a 1 /10 to 1 /20 dilution in extraction buffer. Load duplicate or triplicate wells with each sample. Incubate for 2 to 4 hours at 30 to 37°C or overnight at 4 to 6°C. Wash thoroughly three times with PBST, avoiding cross-contamination of samples.
3. Add unlabelled intermediate antibody at an appropriate dilution, normally 0.25 ?g per ml or less. Incubate for 30 to 60 minutes at 30 to 37°C, and wash plate three times with PBST.
4. Add enzyme-labelled antibody specific to the intermediate antibody, diluted according to the instructions supplied. Incubate 1 to 2 hours at 30 to 37°C and wash plate carefully at least three times with PBST.
5. Add substrate freshly prepared at 0.6 to 1 mg per ml concentration in substrate buffer (10 percent diethanolamine, pH 9.8). Incubate until strong colour change develops in positive controls (normally 30 to 60 minutes) and read plates. Plates may be read at several intervals without stopping the reaction so rate of reaction can be calculated, or the reaction can be stopped at an appropriate time by addition of 3 M NaOH and a single reading can be made. If plates are read visually, score estimated relative strength of reaction. If read on a spectrophotometer or plate reader, record the OD405 values.
Schedule 3
Plate-trapped indirect ELISA
1. Add antigen extracts to uncoated ELISA plates and incubate 1 to 4 hours at 25 to 30°C or overnight at 4 to 6°C. (Note that samples prepared for PTA should not have Tween 20 in the extraction buffer.) Wash plates three times with PBST and avoid contamination while washing.
2. Add unlabelled antibody specific to the antigen at an appropriate dilution (normally 1 ?g per ml or less). Unpurified polyclonal antisera or ascites fluid can be used. Incubate 1 to 2 hours at 30 to 37°C and wash three times with PBST.
3. Add enzyme-labelled antibody specific to the unlabelled antibody, at the specified dilution (normally about 1/1 000), and incubate 1 to 2 hours at 30 to 37°C. Wash carefully at least three times with PBST.
4. Add substrate prepared at a concentration of 0.6 to 1 mg per ml in substrate buffer (10 percent diethanolamine, pH 9.8). Incubate until a strong colour change develops in positive controls (normally 30 to 60 minutes) and read plates. Plates may be read at several intervals without stopping the reaction so rate of reaction can be calculated, or the reaction can be stopped at an appropriate time by addition of 3 M NaOH and a single reading can be made. If plates are read visually, score estimated relative strength of reaction. If read on a spectrophotometer or plate reader, record the OD405 values.
TROUBLE-SHOOTING
Several types of problems may be encountered with ELISA. Some understanding of the operating principles of ELISA helps for systematic troubleshooting to identify and correct the problem. Several of the most common situations are covered here. If the suggestions given do not solve the problem encountered, seek the help of someone who has extensive experience with ELISA.
No reaction or reaction is very slow
The common causes are:
Test conjugate and substrate by mixing a small amount of dilute conjugate with fresh substrate in a small beaker. If no reaction occurs, test each separately again and replace the faulty component. Test reactivity of the antibody by an alternative procedure such as immunodiffusion or microprecipitation. Check calculation of dilutions and test a freshly prepared positive control. Test other extraction buffers. Run a different virus system with the same buffers and protocols.
Colour development is non-specific
When all wells, including buffer and healthy controls, show a strong reaction, it could indicate:
If the buffer control is negative, but the healthy control shows a positive reaction, the antiserum used probably has a high concentration of antibodies to healthy plant antigens. The alternatives are either to absorb the antiserum with healthy plant proteins to remove the antibodies in the serum specific to the healthy plant proteins, or to prepare other antisera.
Colour development is erratic
The common causes of erratic reaction within a plate are:
Check another source of plates and review the care used in the operating procedure.
Reaction very rapid; some reaction also in healthy samples
This normally indicates that the conjugate concentration is much too high. Try several tenfold dilutions. If differentiation still fails to occur between healthy and positive samples with normal incubation periods, see recommendations above.
REFERENCES
Hundreds of references are available on the ELISA technique and its application to numerous plant viruses. We list a few here. Many of these citations contain additional literature citations.
Adams, A.N. & Barbara, D.J. 1982. The use of F(ab'),-based ELISA to detect serological relationships among carlaviruses. Ann. Appl. Biol., 101: 495-500.
Bar-Joseph, M. & Garnsey, S.M. 1981. Enzymelinked immunosorbent assay (ELISA): principles and application for diagnosis of plant viruses. In K. Maramorosch & K.F. Harris, eds, Plant diseases and vectars: ecology and epidemiology, p. 35-59. New York, Academic Press.
Bar-Joseph, M., Garnsey, S.M., Gonsalves, D., Moscovitz, M., Purcifull, D.E., Clark, M.F. & Loebenstein, G. 1979. The use of enzyme-linked immunosorbent assay for detection of citrus tristeza virus. Phytopathology, 69: 190194.
Clark, M.F. 1981. Immunosorbent assays in plant pathology. Annu. Rev. Phytopathol., 19: 83-106.
Clark, M.F. & Adams, A.N. 1977. Characteristics of the micro-plate method of enzyme-linked immunosorbent assay for the detection of plant viruses. J. Gen. Virol., 34: 475-483.
Clark, M.F. & Bar-Joseph, M. 1984. Enzyme immunosorbent assays in plant virology. Methods Virol., 7: 51-85.
Clark, M.F., Lister, R.M. & Bar-Joseph, M. 1988. ELISA techniques. In A. Weisbach & H. Weisbach, eds, Methods for plant molecular biology, p. 507530. New York, Academic Press.
Engelbrecht, D.J. 1980. Indexing grapevines for grapevine fanleaf virus by enzyme-linked immunosorbent assay. Proc. 7th Meet. ICVG, Niagara Falls, NY, USA, 1980, p. 277-282.
Engvall, E. & Pesce, A.J. 1978. Quantitative enzyme immunoassay. London, Blackwell Scientific Publications. 129 pp.
Gonsalves, D. 1979. Detection of tomato ringspot virus in grapevines: a comparison of Chenopodium quinoa and enzyme-linked immunosorbent assay (ELISA). Plant Dis. Rep., 63: 962-965.
Hampton, R., Ball, E. & De Boer, S., eds. 1990. Serological methods for detection and identification of viral and bacterial plant pathogens. A laboratory manual. St Paul, MN, USA, Am. Phytopathol. Soc. Press. 389 pp.
Huss, B., Muller, S., Sommermeyer, G., Walter, B. & Van Regenmortel, M.H.V. 1987. Grapevine fanleaf virus detection in various grapevine organs using polyclonal and monoclonal antibodies. Vitis, 25: /78-188.
Jones, R.A.C. & Torrance, L. 1986. Developments and applications in virus testing. Wellesbourne, War., UK, AAB. 300 pp.
Koenig, R. & Paul, H.L. 1982. Variants of ELISA in plant virus diagnosis. J. Virol. Methods, 5: 113-125.
Kölber, M., Beczner, L., Pacsa, S. & Lehoczky, J. 1985. Detection of grapevine chrome mosaic virus in field-grown vines by ELISA. Phytopathol. Mediterr., 24: 135-140.
Maggie, E.T. 1980. Enzyme-immunoassay. Boca Raton, Florida, USA, CRC Press. 295 pp.
McLaughlin, M.R., Barnett, O.W., Burrows, P.M. & Baum, R.H. 1981. Improved ELISA conditions for detection of plant viruses. J. Virol. Methods, 3: 1325.
Permar, T.A., Garnsey, S.M., Gumpf, D.J. & Lee, R.F. 1988. A monoclonal antibody which discriminates strains of citrus tristeza virus. Phytopathology, 78: 1559.
Sanchez-Vizcaino, J.M. & Cambra Alvarez, M. 1987. Enzyme immunoassay techniques, ELISA, in animal and plant diseases. Tech. Ser. No. 7, 2nd ed. Paris, Office international des epizooties. 54 pp. (Available in English, French and Spanish)
Tanne, E. 1980. The use of ELISA for the detection of some nepoviruses in grapevines. Proc. 7th Meet. ICVG, Niagara Falls, NY, USA, 1980, p. 293-296.
Van Regenmortel, M.H.V. 1982. Serology used immunochemistry of plant viruses. New York, Academic Press. 302 pp.
Vela, C., Cambra, M., Cortes, E., Moreno, P., Miguet, S.G., Perez de San Roman, C. & Sanz, A. 1986. Production and characterization of monoclonal antibodies specific for citrus tristeza virus and their use in diagnosis. J. Gen. Virol., 67: 91-96.
Walter, B. & Etienne, L. 1987. Detection of the grapevine fanleaf virus away from the period of vegetation. J. Phytopathol., 120: 355-364.
Walter, B., Vuittenez, A. Kuszala, A., Stocky, G., Buckard, J. & Van Regenmortel, M. H. V. 1984. Detection sérologique du virus du courtnoué dans la vigne par le test ELISA. Agronomie, 4: 527-534.
FIGURE 262 Diagram of the components of four popular types of ELISA:
a) Double antibody sandwich ELISA (DAS). The most widely used form of ELISA for plant pathogens. The wells of the ELISA plate (the solid phase or immunosorbent surface) are coated with an unlabelled antibody specific to the pathogen, which becomes the trapping antibody (TA). The antigen (V) is captured by the trapping antibody and detected by the enzyme-labelled antibodies (LA), which are normally from the same polyclonal antiserum used for trapping and detection;
b) Double antibody sandwich indirect ELISA (DAS-I). The intermediate antibody (IA) is unlabelled and must be from a different animal species than the coating antibody. The LA is an antibody specific for the IA. It the F(ab')2 antibody component is used for coating, the whole unlabelled antibody from the same animal can be used as the IA and is detected with protein A conjugated to an enzyme;
c) Enhanced DAS-I. This is similar to DAS-I, but the enzyme concentration on the LA is amplified by an additional treatment to increase sensitivity. Frequently, the LA is biotinylated to react to avidin enzyme conjugates; d) Plate-trapped antigen indirect ELISA. The antigen (V) is trapped directly on the plate surface and detected by using an unlabelled antibody specific to the antigen (IA) plus an enzyme-labelled antibody (LA} specific to the IA. Enhancement as shown for DAS-I is also possible
FIGURE 279 Data sheet for recording ELISA results
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}