![]()
![]()
![]()
First draft prepared by
Dr. B. L. Marshall
New Zealand Embassy
Washington DC., USA
ADDENDUM
to the Spiramycin residue monograph prepared by the 43rd meeting of the Committee and published in FAO Food and Nutrition Paper 41/7, Rome 1995
Introduction
As the sponsor was unable to provide a validated chemical method for the analysis of spiramycin and neospiramycin residues in pig tissues to the 43rd JECFA meeting in 1994, it was not possible to estimate the contribution that these residues would make to total residues, and as such the Committee requested that the sponsor provide the following information for consideration by the 47th JECFA meeting in 1996.
1. A validated analytical method for spiramycin and neospiramycin in pig tissues.2. Residue data to estimate the percentage of the total antimicrobial activity represented by spiramycin and neospiramycin in pig liver, kidney and fat.
RESIDUES IN FOOD AND THEIR EVALUATION
Methods of Analysis for Residues in Tissues
In response to the Committee's request, the sponsor provided performance validation data from a number of authors, to support microbiological agar gel diffusion methods for spiramycin determination. (Pascal et al., 1990b; Cuypers et al., 1994; and Daix and Gougeard, 1996) The sponsor also reviewed two HPLC methods for spiramycin determination in pig liver (Mourier, et. al., 1993; and applied by Cuypers et al., 1994) and in pig muscle and liver tissues (National Agency of Veterinary Medicine - CNEVA, Fougères, France, 1993, that has been implemented by CEPHAC Research Centre, Saint Benoit, France). Reference was also made by the sponsor, to a third HPLC method for pig muscle and liver tissues (Mignot, Lefebre and Millerioux., 1993) but the data was not presented because the results were determined to be ambiguous, unclear and not consistent with those obtained using the microbiological assay method accepted at the 38th JECFA meeting. Not only did the authors record chromatographic interference but the analytical results were apparently significantly lower than those obtained by the microbiological assay. A further HPLC method deemed appropriate by Danish Authorities, for the determination of spiramycin and tylosin residues in pig muscle tissue, was also received for consideration, from Nielson et al., (1995) of the Danish State Veterinary Laboratory.
Data was provided by the sponsors to support the recommendation that the microbiological gel diffusion assay for the determination of spiramycin and its active metabolites, that was developed by Pascal et al. in 1989, submitted to JECFA in 1990, recognised in the European Pharmacopoeia, and further validated by Cuyper et al., 1994, and Daix and Gougeard, 1996, is more appropriate for routine monitoring of pig tissues than HPLC analysis. Comparative studies of the HPLC and microbiological methods by Cuypers et al., confirmed excellent correlation between the two.
The microbiological diffusion assay was performed according to the criteria described in the European Pharmacopoeia, II edition, using Micrococcus luteus ATCC 9341 as the test strain and medium A (without pancreatic digest of casein) as culture medium. Ground liver samples were extracted three times with a mixture of methanol and pH 9 phosphate buffer, 7:3. After evaporation of the methanol and adjustment of pH, the solutions were deposited on inoculated culture media, incubated at 30°C for 36 hours, zones of inhibition read and sample titers calculated.
Validation of Analytical Methods
Microbiological Methods
The Pascal et al., 1990b, residue feeding study was designed to determine the kinetics of spiramycin elimination in muscle, liver, kidney and fat tissues of sixty-six Large White X French Landrace male and female piglets, approximately 11 weeks of age and weighing 25-30 kg, which had been fed daily for 7 days, medicated feed containing 16 or 25 mg/kg BW spiramycin embonate (WHO standard 3200 IU/mg). To determine possible interference on the elimination kinetics of spiramycin treated animals, from concomitant use of oxytetracycline (OTC) or sulphamethazine (SMZ), two of the animal groups, each of 12 animals, were also fed feed containing 32 mg/kg BW OTC or 32 mg/kg BW SMZ. The absence of interference with OTC or SMZ was evaluated by incorporating spiramycin in blended liver or kidney tissues with twice as much OTC or SMZ.
At each evaluation time, 2-4 piglets were slaughtered, and six 8-10 g samples of each edible tissue taken for analysis. After solvent extraction and clean-up, spiramycin was assayed by agar diffusion, using Micrococcus luteus ATCC 9341 as the test organism. Reference spiramycin solutions were prepared using spiked tissue. This microbiological assay method was validated in terms of linearity, parallelism; extraction yield; sensitivity of titration; and repeatability.
The analytical parameters of the method, including extraction yields, assay sensitivity and repeatability and the mean coefficient of correlation of liver, kidney and muscle test samples (from triplicate, quintuplicate and a single test sample) are summarised in Table 1.
Table 1. Analytical Parameters of the Pascal et al 1989 Microbiological Assay in Pigs
|
Tissue |
Coefficient of correlation |
Extraction yield (%) |
Sensitivity (LOD) (m g/kg) |
Repeatability (%) |
|
Liver |
0.945 |
at 3000 m g/kg: 80 |
300 |
1.97 (SD = 0.22) |
|
at 6000 m g/kg: 84 |
||||
|
Kidney |
0.940 |
at 1200 m g/kg: 83 |
150 |
1.12 (SD = 0.08) |
|
at 1600 m g/kg: 79 |
||||
|
at 8000 m g/kg: 89 |
||||
|
Muscle |
0.946 |
at 500 m g/kg: 90 |
100 |
|
|
Fat |
0.629 |
at 500 m g/kg: 69 |
100 |
|
While the performance standards were generally very acceptable, the coefficient of correlation and the extraction yield for fat was poor. This undoubtedly is due to the greater physical heterogeneity of the tissue and hence the difficulty in homogenising and extracting the residues.
In the study, tissue concentrations in animals receiving 16 mg/kg BW/d spiramycin, rapidly decreased after treatment ceased, regardless of whether spiramycin had been administered in feed separately or in conjunction with OTC or SMZ. The data shows that muscle residue concentrations fell to 120 m g/kg within 12 hours of cessation of treatment and were at or below the limit of detection by day 3. By day 10 liver and kidney concentrations were below the detection limit of the method, 300 and 150 m g/kg, respectively. For animals receiving 25 mg/kg BW/d, liver and kidney concentrations were below the detection limit on day 20, with muscle and fat concentrations being below the limit of detection regardless of the sampling time frame (7,10 and 20 days post-treatment). Spiramycin residue concentrations in fat were found to be consistently below the detection limit (100 m g/kg) of the method at the both dose levels.
The antimicrobial activity of neospiramycin was determined to be closely equivalent to that of spiramycin.
Table 2 summarises the mean triplicate spiramycin concentrations (m g/kg) in liver and kidney tissue samples determined by microbiological assay from piglets fed 16 mg/kg BW spiramycin alone or in conjunction with OTC or SMZ fed at 32 mg/kg BW/d for 7 days.
Table 2. Absence of Interference by OTC or SMZ on the Elimination Kinetics of Spiramycin in Treated Piglets
|
Tissue |
Liver (m g/kg) |
Kidney (m g/kg) | ||||
|
Withdrawal (days) |
Spir |
Spir + OTC |
Spir + SMZ |
Spir |
Spir + OTC |
Spir + SMZ |
|
0 |
6270 |
|
|
8930 |
|
|
|
3 |
1430 |
|
|
1280 |
|
|
|
7 |
580 |
640 |
430 |
210 |
240 |
170 |
|
10 |
< 300 |
380 |
< 300 |
< 150 |
< 150 |
< 150 |
|
15 |
< 300 |
< 300 |
< 300 |
< 150 |
< 150 |
< 150 |
|
20 |
< 300 |
< 300 |
< 300 |
< 150 |
< 150 |
< 150 |
Spir - spiramycin; OTC - oxytetracycline; SMZ - sulphamethazine (sulphadimidine)
Mean spiramycin tissue residue recovery values for liver and kidneys are summarised in Table 3.
Table 3. Mean Spiramycin Tissue Recovery Values, Expressed as a Percent with Respect to the Addition of Spiramycin Alone
|
Tissue |
Spiramycin + OTC |
Spiramycin + SMZ |
|
Liver |
111 (SD = 5.2) |
95 (SD = 4.2) |
|
Kidney |
93 (SD = 12.2) |
102 (SD = 6.6) |
Data presented demonstrated that in a study to determine the sensitivity of M. luteus ATCC 9341 to OTC alone, no antibiotic activity was evident at concentrations ranging from 50 to 500 m g/l. Similarly there was no significant interference of OTC evident in the assay for spiramycin, although there was a little synergy, either with or without extraction, when OTC was added at twice the concentration of spiramycin.
Table 4 indicates the % recovery with or without extraction, when spiramycin was present in concentrations of 44 to 500 m g/l and OTC, 88 to 1000 m g/l.
Table 4. Absence of Interference of OTC in the Determination of Spiramycin With* or Without** Extraction. Concentration OTC = 2 x Concentration of Spiramycin
|
|
Spiramycin |
OTC |
Spiramycin + OTC |
|
% Recovery** |
100 |
0 |
108 |
|
% Recovery* |
100 |
0 |
112 |
There is little or no change in the elimination kinetics of Spiramycin in animals where oxytetracycline or sulphamethazine has been administered in combination with Spiramycin, as compared to animals dosed with Spiramycin alone and at the same dose rate. Given the precision of this method, it can be concluded that with little interference from OTC or SMZ, this microbiological assay would be suitable for routine monitoring of Spiramycin residues in pig tissues.
Another study reported by Daix and Gougeard., 1996, further defined the validation parameters (limits of detection, quantification and repeatability) of the Pascal et al., 1990b microbiological agar diffusion method for pig liver, kidney, muscle and fat tissues and demonstrated the suitability of this method for routine monitoring of pig tissues. Limits of detection and quantification were determined by testing 3 replicates, at each of the 3 concentrations of incurred residues. This involved 1/3 dilutions of the control extracts that had been prepared by adding a quantity of reference Spiramycin to 10 g of homogenised tissue. The estimated repeatability was based on testing 6 replicates of each tissue of liver, kidney, muscle and fat, at 2 x MRL, as allocated by the 43rd JECFA, for the corresponding tissue, ie at concentrations corresponding to 2 x 600 m g/kg in liver; 2 x 300 m g/kg in kidney; 2 x 200 m g/kg in muscle; 2 x 200 m g/kg in fat. The relative standard deviation for the inhibition zone was found to be less than 5% for all tissues. Results of this study are summarised in Table 5.
Table 5. Detection, Quantification and Repeatability Validation Parameters for the Spiramycin Microbiological Assay of Different Pig Tissues
|
Tissue |
Limit of Detection (m g/kg) |
Limit of Quantification (m g/kg) |
Repeatability (%) |
|
Liver |
140 |
300 |
2.9 |
|
Kidney |
140 |
300 |
2.9 |
|
Muscle |
45 |
100 |
2.5 |
|
Fat |
70 |
115 |
2.2 |
It was concluded that the microbiological diffusion assay developed by Pascal et al., (1990b) and described in report RPS JP/LY ref. 1103 of January 1990, has a satisfactory repeatability in the four tissues studied and at concentrations consistent with practical conditions.
Chemical Methods (HPLC)
An HPLC method developed by the National Agency of Veterinary Medicine - CNEVA, Fougères, France, for the determination of Spiramycin and neospiramycin in cattle tissues and modified by Mignot et al (1993), was evaluated as to suitability for routine screening or as a reference method, for pig liver and muscle tissues. Both Spiramycin and neospiramycin were extracted from tissues by liquid-liquid extraction, followed by solid-liquid phase extraction. The eluates were chromatographed using reverse phase high performance liquid chromatography (HPLC) with an acidic mobile phase and UV detection at 231 nm. The method was fully validated in muscle for spiramycin and neospiramycin, in terms of linearity, recovery, with intraday and interday (frozen muscle samples) precision and accuracy being considered acceptable. CV and % error were both < 20 %, and the LOQ was validated at 25 m g/kg for both compounds. The mean recovery of the associated quality control freshly prepared samples was equal to 93 % for spiramycin and 100% for neospiramycin. Only 4 out of 16 samples however, had residue concentrations above the limit of quantification. While the liver tissue assay was validated in terms of linearity and intraday precision and accuracy, the recovery was < 50%, and the interday precision and accuracy could not be determined. Although spiramycin and neospiramycin residues were quantified in liver tissue samples with incurred residues, and a mean recovery of 83 % for spiramycin and 80% for neospiramycin determined, residue concentrations above the limits of quantification 200 m g/kg for spiramycin and 100 m g/kg for neospiramycin were measured in only 5 out of 16 samples. Due to chromatographic interferences in liver and residues invariably being below the limit of quantification in muscle and liver tissues, it was concluded that this procedure would not be suitable for routine analysis of pig muscle and liver tissues.
An HPLC method, developed by the Food Control Laboratory of the Danish Veterinary Service (Petersen, et al., 1995) and currently used in Denmark to determine spiramycin and tylosin residues in muscle tissue, was forwarded to JECFA for consideration. The data demonstrated that while the method is suitable for muscle tissue as a screening or reference method, it is unsuitable for kidney or liver tissues. While plasma analysis is considered possible, the laboratory was unable to provide documentary evidence on the ratio distribution between plasma and muscle tissue. The results of 16 blind pig muscle samples used to determine detection (LOD) and quantification (LOQ) limits for spiramycin and tylosin are shown in Table 6.
Table 6. LOD and LOQ of Spiramycin and Tylosin in Pig Muscle
|
Analyte |
LOD (m g/kg) |
LOQ (m g/kg) |
|
Spiramycin |
18 |
33 |
|
Tylosin |
27 |
40 |
Repeatability and reproducibility, done on spiked standard curves run twice a day over six days, are shown to be high at 0 m g/kg for spiramycin and tylosin. The reason is, that noise on the base line varies both under clean-up at day one, and between days. All peaks were integrated using the same method of integration, with peaks with 0 m g/kg values, not being subject to forced integration. A low pH was found to be necessary during optimisation of the extraction process in order to obtain a good recovery for spiramycin, however, a low pH could give a lower recovery for tylosin. An assessment of the robustness and extraction process showed that while the quantity of solvent used was significant on the percentage recovery of spiramycin, interactions were not found to be significant.
From the data provided, not only do liver concentrations also need to be considered cautiously, because of chromatographic interferences, but spiramycin and neospiramycin concentrations in pig liver and muscle are often below the limit of quantification. The method should only be considered useful for determining the total spiramycin and tylosin residue concentrations in muscle tissues.
Percentage Total Antimicrobial Activity Represented by Spiramycin and Neospiramycin in Pig Liver
An HPLC method, developed by Mourier (1993), and applied by Cuypers et al (1994), was used in a study, in conjunction with a microbiological assay, to determine the metabolism of spiramycin (embolate) in liver tissues of 12 treated pigs, fed 22 mg/kg bw/d spiramycin in feed for 7 days, 4 of which were slaughtered at each experimental withdrawal time points of zero, 3 and 10 days. The percentage of total activity represented by spiramycin and neospiramycin in pig liver was also determined. The method incorporates an internal standard RP 22711, a column temperature set at 60°C and involves extraction with acetonitrile: water (90:10, v/v), which can detect transformed spiramycin down to 200 m g/kg. Concentration of the extract on a solid phase extraction precolumn occurs before separation and HPLC analysis using UV detection at 232 nm, the absorption maximum of spiramycin.
Table 7 indicates the extraction liters data obtained from Mourier's acetonitrile/water extraction, incurred residue control study. Spiramycin I and III accounted for 400 m g/kg out of the total antimicrobial activity of 13800 m g/kg. The extraction titer for the internal standard was 21500 m g/kg. The percentage of total microbial activity represented by spiramycin (1 and III) and neospiramycin in this incurred pig liver study was calculated to be 2.8%.
Table 7. Extraction Titers (m g/kg) of Spiramycin and Its Metabolites from Treated Pig Liver
|
|
tr Neospira I |
tr Spira I |
Spira I |
tr Neospira III |
tr Spira III |
Spira III |
|
(m g/kg) |
1400 |
6600 |
200 |
900 |
4500 |
200 |
tr = cysteine conjugate; Neospira = neospiramycin; Spira = spiramycin
As the first extraction is not complete, an underestimation could occur of the quantity of transformed spiramycin and transformed neospiramycin because of their higher polarity.
The report of Cuypers et al., (1994) demonstrates (Table 8) mean spiramycin residues levels (m g/kg) in liver tissue of pigs fed daily 22 mg/kg bw spiramycin medicated feed for 7 days.
Table 8. Mean Spiramycin Metabolite Residue Levels (m g/kg) in Liver Tissue of Pigs Fed 22 mg/kg/d Spiramycin Medicated Feed for 7 Days
|
Withdrawal (days) |
Spiramycin metabolites (m g/kg)* |
|||||
|
tr Neospira I |
tr Spira I |
Neospira I |
tr Neospira III + Spira I (coelution) |
tr Spira III |
Spira III |
|
|
0 |
200 |
1800 |
100 |
1000 |
1800 |
200 |
|
3 |
ND |
80 |
130 |
430 |
280 |
ND |
|
10 |
ND |
ND |
30 |
ND |
80 |
ND |
*Mean of four pigs; results expressed in m g/kg calculated with reference to internal standard RP 22711; ND = Not Detected; limit of quantification 100 m g/kg
The conclusion of these studies was that the parent drug spiramycin, which consists of two major components, spiramycin (I and III), is found in liver extracts of orally treated pig, in the three forms, L-cysteine transformed spiramycin and neospiramycin, and non transformed spiramycin base, generally present in very small quantities. The transformed/non-transformed base ratio is never found to be less than 0.9, and depends above all, on the quantity of the L-cysteine present in the extract. The ratio of Spiramycin ILiver (tr spiramycin I + tr neospiramycin I+ spiramycin I)/Spiramycin IIILiver was calculated to be around 1.5, about ten times lower than the Spiramycin I/Spiramycin III ratio characteristic of spiramycin used for treatment. Spiramycin II, present in feed in negligible quantities was not found in liver in any form whatsoever. The internal standard (RP 22711) is not transformed by L-cysteine, but found in liver in its original form. Control liver showed no trace of spiramycin or its derivatives. Both the repeatability and reproducibility of spiked control liver were considered satisfactory. Table 9 and the 2.5% CV of reproducibility of the internal standard, indicate that extraction and injection are reproducible.
Table 18. Percentage Coefficient of Variation of Repeatability and Reproducibility of Control Spiked Liver
Repeatability
|
|
tr Neospira I |
tr Spira I |
Neospira I |
tr Neospira III + Spira I Coelution |
tr Spira III |
Spira III |
|
CV (%) |
7.3 |
6.5 |
21.3 |
11.7 |
2.6 |
12.2 |
Reproducibility
|
|
tr Neospira I |
tr Spira I |
Neospira I |
tr Neospira III + Spira I Coelution |
tr Spira III |
Spira III |
|
CV (%) |
30.2 |
6.1 |
16.3 |
10.0 |
4.6 |
24.4 |
tr = L-cysteine conjugate
Approximate retention times are provided in Table 10. A chromatograph of a liver extract from a treated pig, demonstrating the complexity involved in quantifying spiramycin residues by HPLC, is provided in Figure 1.
Table 10. Approximate Retention Times (min) of Spiramycin and its Derivatives
|
Solute |
Retention Time Relative to Spiramycin I |
Retention Time (min) |
|
tr Neospiramycin I |
0.56 |
4.3 |
|
tr Spiramycin I |
0.67 |
5.2 |
|
Neospiramycin I |
0.74 |
5.7 |
|
Spiramycin I |
1.00 |
7.7 |
|
tr Spiramycin III |
1.30 |
10.1 |
|
Internal Standard RP 22711 |
1.71 |
13.2 |
|
Spiramycin III |
2.00 |
15.5 |
tr = L-cysteine conjugate
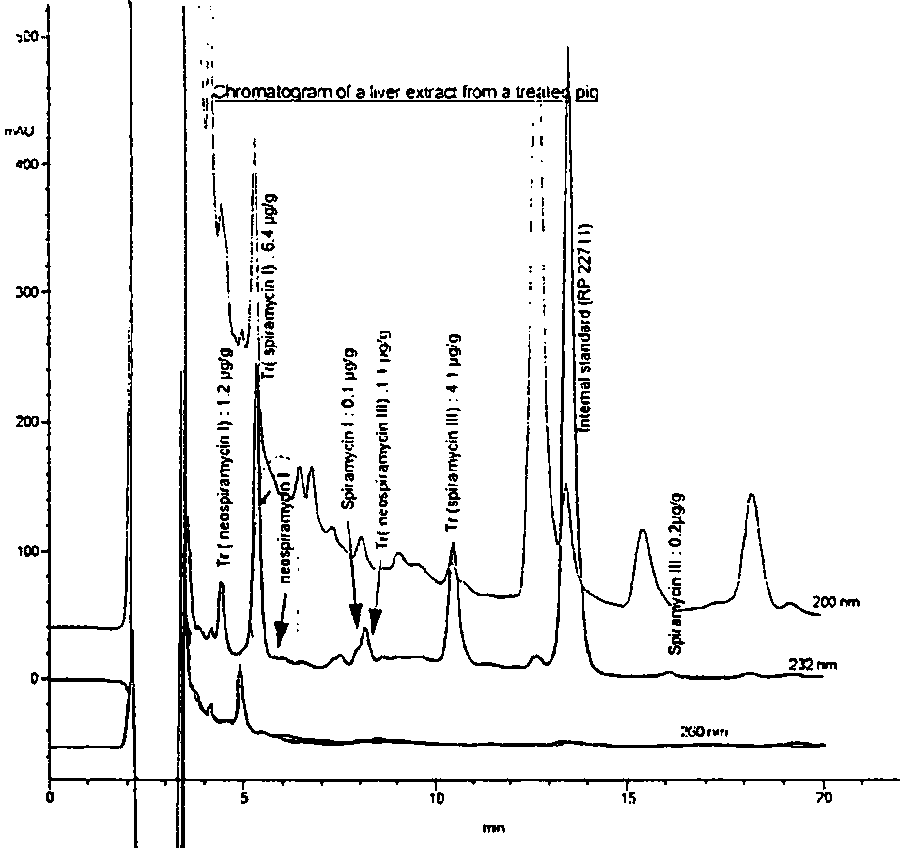
The microbiological M. luteus ATCC 9341 agar gel diffusion assay, described in the European Pharmacopoeia, II Edition and reported in the note of Pascal, 1990, and which was also carried out on the same liver sample as with the HPLC method, showed that there is excellent correlation between the two techniques (Table 11). Microbial values were expressed as spiramycin equivalent. From the data of Mourier et al, 1993, the sum of the metabolites assayed by HPLC were 13800 m g/kg and 12600 m g/kg for the microbial method, proving an excellent correlation.
Table 11. Comparison of Mean Results of HPLC and Microbiological Assays of Quadruplicate Pig Liver Samples
|
Withdrawal Time (days) |
HPLC (m g/kg) |
Microbiological Assay (m g/kg) |
|
0 |
5000 (SD = 1.7) |
5300 (SD = 1.6) |
|
3 |
900 (SD = 0.3) |
1300 (SD = 0.1) |
|
10 |
100 |
200 |
A comparison between spiramycin I, II, and III and neospiramycin I and their combinations with cysteine in Mueller-Hinton agar and broth demonstrated that the minimum inhibitory concentration (MIC) or microbiological activities of cysteine conjugates was found to be somewhat lower than the relevant parent compound, with the percentage being 50-100 %. Besides with tylosin, a degree of antibacterial cross reactivity has been demonstrated to occur in vitro activities against Micrococcus luteus ATCC 9341. (Table 12).
Table 12. Micrococcus luteus Inhibition by Four Antibiotics
|
Antibiotics |
MIC (mg/l) |
|
Amoxicillin |
0.008 |
|
Gentamicin |
1 |
|
Spiramycin |
0.5 |
|
Rifamycin |
0.008 |
APPRAISAL
Spiramycin is a macrolide antibiotic that is produced by certain strains of Streptomyces ambofaciens and used in oral or parenteral formulations for the treatment or prophylaxis of local or systemic diseases in cattle and pigs. It had been previously evaluated at the thirty-eighth and forty-third meetings of the Committee. A validated chemical method had not been available for the analysis of Spiramycin and neospiramycin residues in pig tissues, and as such it had not been possible to estimate the percentage contribution that these residues would make to total residues.
The 43rd Meeting of the Committee required the following information for evaluation in 1996:
1. A validated analytical method for determining the concentrations of Spiramycin and neospiramycin in the edible tissues of pigs.2. Residue data to estimate the percentage of the total antimicrobial activity accounted for by Spiramycin and neospiramycin in the liver, kidney and fat of pigs.
Analytical methods
Microbiological and HPLC methods and respective study data were provided for the determination of Spiramycin and neospiramycin in pig tissues.
Microbiological Methods
A method that was submitted for consideration at the forty-third meeting of the Committee was re-evaluated because additional information on the limit of detection, the limit of quantification and repeatability was provided. The method, which has been published in the European Pharmacopoeia, was accepted at the forty-third meeting of the Committee for screening muscle tissues for evidence of Spiramycin residues. This method uses a solvent extraction step followed by a microbiological gel diffusion assay involving Micrococcus luteus ATCC 9341 as the test strain. The limits of quantification of the method, which were estimated using fortified tissues of a control pig, were 300 m g/kg for liver and kidney, 100 m g/kg for muscle and 115 m g/kg for fat. Recoveries were > 80% for liver, kidney and muscle but were lower (69%) for fat. Using incurred tissues at the recommended MRL, the coefficient of variation of the mean of 2-3 replicate analyses of the same sample was in the order of 20-25 %, with a range less than 5 % to more than 50 %. Analyses of incurred liver tissue with residues slightly above the MRL showed that the results with the microbiological assay were, on average, about 40% higher than those obtained with an HPLC method.
The microbiological method was specific for Spiramycin in the presence of oxytetracycline and sulfadimidine, which may be formulated in feed together with Spiramycin.
The Committee concluded that this microbiological method is suitable to screen pig tissues for residues of Spiramycin and its active metabolites, providing that results could be confirmed with a more specific method.
However, the possibility of cross reactivity with hydrophilic antibiotics cannot be excluded.
Chemical methods
Data submitted from a number of HPLC studies were considered. An HPLC method using fortified samples demonstrated suitable sensitivity (limit of detection of 18 m g/kg, and limit of quantification of 33 m g/kg) for analyzing muscle tissue for spiramycin. Due to chromatographic interferences, it was found to be unsuitable for analysis of kidney or liver tissue. It was also specific in the presence of tylosin.
A further HPLC study using spiked samples demonstrated that the limits of quantification for spiramycin and neospiramycin in muscle samples were 25 m g/kg with recoveries of 93 and 100%, respectively. The limit of quantification for liver was 200 m g/kg for spiramycin and 100 m g/kg for neospiramycin, with recoveries in the range of 83 to 80%, respectively.
There was good correlation between the HPLC method and the microbiological assay as demonstrated in the measurement of incurred liver tissues containing residues in the range 100-5000 m g/kg.
The Committee concluded that there are HPLC methods that were suitable for measuring spiramycin residues in muscle, liver and kidney at the level of the MRL. Tylosin did not interfere in the HPLC assay.
Antimicrobial Activity
In an HPLC study, using tissues from pigs fed spiramycin, the spiramycin and neospiramycin cysteine conjugates were found to account for 97.5 % of the residues, with the cysteine conjugates accounting for approximately 90% of the antimicrobial activity of the parent drug, thereby supporting ths use of the antimicrobial assay for routine screening.
Maximum Residue Limits
To promote method validation the Expert Committee considered it appropriate to harmonize MRLs for parent spiramycin residues in different tissues of different food producing animals. The following MRLs were established:
|
Muscle |
(cattle, pigs, chickens) |
200 m g/kg |
|
Liver |
(cattle, pigs, chickens) |
600 m g/kg |
|
Kidney |
(cattle, pigs) |
300 m g/kg |
|
(chickens) |
800 m g/kg |
|
|
Fat |
(cattle, pigs, chickens) |
300 m g/kg |
|
Milk |
(cattle) |
100 m g/l |
expressed as the sum of spiramycin and neospiramycin for cattle and chickens, and as spiramycin equivalents (antimicrobially active residues) for pig tissues.
The Committee agreed that an MRL for chicken kidney should be recommended at 800 m g/kg. Considering a standard daily intake of 300 g muscle, 100 g liver, 50 g kidney, 50 g fat and 1.5 litres milk, a theoretical maximum daily intake of spiramycin residues will be 440 m g. Using an ADI of 0-50 m g per kg of body weight, a 60-kg person, established at the 43rd meeting of the Committee, would therefore be permitted to consume 3000 m g/kg.
The Committee recommended that the current temporary MRLs for pig liver, kidney, and fat be established as fall MRLs.
REFERENCES
CNEVA - Fougères HPLC method: from "Determination of spiramycin and neospiramycin in biological samples (muscles and liver) from a metabolism study of spiramycin in the pig". Report CEPHAC No. CD 514, April 1993.
Cuypers, V., Mourier, P., Poirier, J. and Rombi, J. (1994). Spiramycin embonate (RP 5337, Embonate). Study of metabolism in pigs. Rhône-Poulenc Rorer Report RPR/RD/CRVA/AN No. 7015, 19 January 1994.
Daix, V. and Gougeard, N. (1996). Spiramycin assay in swine tissues complement of validation of the microbiological assay method. Report RPR/RD/CPD/PQA/CRVA 7370, 15 May 1996.
Ferriot, A. and Videau, D. (1971). Elimination et fixation tissulaire de la Spiramycine chez le porc. Can. Med. Vet., 40, 164-169, 1971.
Huet, A.M., Bosc, F. and Floch, R. (1988). Procedure for the determination of spiramycin in the serum or plasma by microbiological method of diffusion on agar. Rhône-Mérieux Report No. MET Oil. Submitted by Rhône-Poulenc Santé, Vitry-sur-Seine, France.
May & Baker (1967). Huntington Research Center, August 1967. Submitted by Rhône-Poulenc Santé, Vitry-sur-Seine, France.
Mignot, A., Lefebre, M.A. and Millerioux, L. (1993). Determination of spiramycin and neospiramycin in biological samples (muscle and liver) from a metabolism study of spiramycin in the pig. CEPHAC Report No. CD 514, April 1993. (p. 44-132).
Mourier, P. (1993). Spiramycin embonate in pigs: study of metabolism. Submitted by Rhône-Poulenc Rorer Report RPR/RD/CRVA/AN No. 6993, 8 December 1993, p. 133-168.
Mourot, D. and Sanders, P. (1988). Determination of spiramycin in the plasma by high performance liquid chromatography. (Ref. Rhône-Mérieux: MET 008) National Veterinary Drugs Laboratory (Fougères). Submitted by Rhône-Poulenc Santé, Vitry-sur-Seine, France.
Nielsen, M.B., Rasmussen, A., Petersen, I., Ørntoft, I. and Petersen, H.W. (1995). Determination of spiramycin and tylosin in muscle - Documentation. Report Submitted by the Ministry of Agriculture and Fisheries, Danish Veterinary Services, Food Control Laboratory, September 1995.
Pascal, C., Bertrand, A., Kies, A. and Poirier, J. (1990b). Spiramycin - Study of residues in swine following administration in feed. Unpublished Report No. 1103. Submitted by Rhône-Poulenc Santé, Vitry-sur-Seine, France, 11 January 1990, p. 1-42.
![]()
![]()
![]()