![]()
![]()
![]()
Reliable data on the nutrient composition of foods can only be obtained by the careful performance of appropriate, accurate analytical methods in the hands of trained analysts. The choice of the appropriate methods carried out under quality assurance schemes is the second crucial element in ensuring the quality of the values in a food composition database.
For many nutrients, several alternative analytical methods are available that, it is often assumed, give comparable results. In fact, methods vary in their suitability for a given analysis and different food matrices. Before the relative merits of particular methods are discussed in Chapter 7 it is necessary to consider the principles involved in method selection. In doing so it is recognized that the analysts' choices may be limited by the resources available; this makes it all the more important to understand the principles involved in method evaluation, particularly the need to define the limitations of any given method.
The evaluation of methods is not the purview of the analysts alone. The technical and scientific advisers to the database programme should be thoroughly conversant with the underlying principles of analytical methodology and the various methods themselves, sharing the responsibility with the analyst for choosing a method.
Compilers should also endeavour to be knowledgeable about the analytical methods used. They are responsible for scrutinizing methods when assessing non-commissioned data or published analyses to assess their suitability for inclusion in the database and to devise the specification for contracts for the preparation of sampling and analytical protocols.
It is also desirable that the professional users of a database should have some understanding of the analytical methods used, and that specialist users should be conversant with the methods used for the nutrient(s) relating to their special interests.
At present there are a number of methodological limitations in the production of data for certain nutrients. Based on a review of methods, Stewart prepared a table summarizing the position in 1980 and 1981, which was later extended by Beecher and Vanderslice (1984). In the table the nutrients were grouped according to the availability of valid methods to measure them. The expanded interest in nutrient composition in legislation and for use in epidemiological research has resulted in further work on method evaluation and development. In the United States, the Association of Analytical Communities (AOAC International) carried
|
Table 6.1 Availability of methods for nutrient analysis (adequacy of methods) | ||||
| Nutrient | Good | Adequate | Not adequate for certain foods |
Lacking |
Moisture |
Moisture |
|
|
|
Nitrogenous constituents |
Total nitrogen, amino acids |
|
Protein, non-protein nitrogen |
|
|
Lipid constituents |
Fatty acids |
Cholesterol, phospholipids, trans fatty acids, individual triacylglycerols |
Some isomeric fatty acids |
|
|
Carbohydrates and dietary fibre |
Individual sugars, starch, non-starch polysaccharides |
Totaldietary fibre, individual non-starch polysaccharides, resistant starch |
|
Lignin |
|
Inorganic nutrients |
Sodium, potassium, calcium, magnesium, phosphorus, iron, copper, zinc, boron, chloride |
Selenium, manganese, fluorine |
Chromium, haem iron, cobalt, molybdenum |
|
|
Vitamins |
Thiamin, riboflavin, niacin |
Vitamin C, retinol, carotenoids, vitamin E, vitamin D, vitamin B6,total folates, folic acid, biotin, pantothenic acid, vitamin B12 |
Some carotenoid isomers, vitamin K |
Some folate isomers |
out a review of methods for use in nutrition legislation (Sullivan and Carpenter, 1993), and major reviews of micronutrient methods were undertaken by the Food Standards Agency in the United Kingdom (2002).
Studies for the development of standard reference materials (SRMs) undertaken in the United States by the National Institute of Standards and Technology (NIST) and in Europe by the Community Bureau of Reference (BCR) have also contributed to method development.
Stewart's original assessments have been updated in Table 6.1, which presents a revised version based on a review undertaken to assess the compatibility of methods (Deharveng et al., 1999). In the table, “good” methods have been extensively evaluated in collaborative trials, “adequate” methods have been subjected to more limited study, and methods categorized as “not adequate for certain foods” have not been studied on a wide range of food matrices. It is important to note that these assessments hold true only when the analyses are carried out by trained analysts and that they do not include any consideration of speed or costs.
The table does not include the wide range of biologically active constituents that are now considered as candidates for inclusion in food composition databases. The methodologies for most of these constituents have not yet been widely studied in collaborative trials.
The primary objective of food composition databases is to provide their users with compositional information on nutrients; therefore the primary factor in the choice of methods is the appropriateness of the analysis in terms of providing the information required by the users. The measurements must provide values that can be used to assess the nutritional value of foods. This means that the database users' requirements may differ from those concerned with the regulation of food composition or the quality control of food in production. Thus, while the measurement of crude protein (total nitrogen multiplied by a factor) is adequate for many purposes, amino acid data would provide a better assessment of the nutritional value of a food. A value for total lipids may be adequate in relation to food quality control, whereas a nutritionist would require assessments of triacylglycerols, sterols and phospholipids separately and detailed fatty acid data. Similarly, while total carbohydrate values may be adequate for food quality control, a nutritionist would require specific values for the different carbohydrates (FAO/WHO, 1998). As a consequence, more biochemically orientated methods are often required when obtaining values for food composition databases.
In some countries, the choice of method may be prescribed by national legislation. In other countries, the regulations often permit the use of methods that give comparable, i.e. similar, values to those obtained by the official methods.
Other considerations will also influence the choice of method. The use of some of the most advanced methods may require substantial capital investment to provide the necessary instrumentation. Considerable resources are also required in the form of trained staff to operate and maintain the instrumentation. The development of such instrumental methods represents a preference for investing in capital rather than in recurrent staff costs and for reducing the cost per analysis by speeding up analysis.
It is incorrect to give the impression that nutrient analyses cannot be performed without such sophisticated instrumentation; for many nutrients classical manual methods are available that give equally sound values. These methods are labour-intensive rather than capital-intensive.
It is true that analyses of certain nutrients, fatty acids for example, do require instrumentation; where this is lacking a laboratory would need to seek collaborative arrangements to acquire the data.
Laboratories in developing countries may lack funds for capital outlay (especially as foreign currency) and lack the resources for the specialized maintenance and supplies necessary for high-technology instrumentation. On the other hand, local funds may be available for technical staff with the necessary background for carrying out non-instrumental methods that provide valid data. A comprehensive range of compatible methods has therefore been covered in Chapter 7.
Laboratories should focus their attention on evaluating and improving the quality and performance of the methods currently employed rather than attempting to institute a wide range of methods using new, untried, methods or losing confidence because of their lack of sophisticated equipment. In many cases, implementing a data quality assurance system and training staff are often better ways to produce good-quality compositional data.
The formal training of food analysts, where it is carried out, usually focuses on the highly accurate detection of compounds appropriate for food regulations. These compounds are often contaminants, which are present at low levels, and the choice of method generally emphasizes levels of detection, sensitivity and precision. In nutrient analysis for a food composition database, the requirements for accuracy and precision may be orientated more towards the recommended intake of a nutrient and the relative importance of the food being analysed in the diet (Stewart, 1980). Analysts may, for example, spend considerable effort measuring vitamins in foods at levels that are nutritionally insignificant.
This difference in emphasis underlines the need for all individuals involved in producing data to be familiar with the objectives of the work, from sampling through to analysis. Sampling protocols should specify the levels of accuracy that are expected. It is also important to maintain a regular dialogue between compilers and the sampling and analytical teams throughout the duration of the work.
While the appropriateness of the method may be a primary factor in method selection, it is also necessary to take into account the analytical attributes of the method.
It is useful to consider a number of points suggested by Egan (1974):
The analytical method selected also needs to have adequate performance characteristics. Büttner et al. (1975) summarize these as reliability criteria (specificity, accuracy, precision and sensitivity) and practicability criteria (speed, costs, technical skill requirements, dependability and laboratory safety).
Thus “reliability” represents a summation of the more conventional measures of method performance. Many analysts would also consider another attribute as falling within this summation: “robustness” or “ruggedness”. This attribute is described below.
(adapted from Horwitz et al. [1978], with permission)
This is a qualitative term expressing a degree of satisfaction with the performance of a method in relation to applicability, specificity, accuracy, precision, detectability and sensitivity, as defined below, and is a composite concept (Egan, 1977). It represents a summation of the measurable attributes of performance. The analyte and the purposes for which the analyses are being made determine the relative importance of the different attributes. Clearly, the analysis of a major constituent such as protein, fat or carbohydrate in foods does not require the same low limit of detection as that needed for the measurement of a carcinogenic contaminant. Conversely, the measurement of a constituent at low levels in foods (e.g. most trace elements, selenium, chromium or vitamins such as vitamin D, vitamin B12 and folates) cannot be expected to deliver the same high accuracy or precision as found with the major constituents.
Horwitz, Kamps and Boyer (1980) found from a study of the results of a large number of collaborative studies undertaken under the auspices of the AOAC that there was a strong empirical relationship between the concentration of an analyte and the observed precision obtained by experienced analysts. The relationship they found was:
CV = 2(1 – 0.5 logC)
where CV is the coefficient of variation and C the concentration g/g.
Many workers use this relationship when assessing the performance of methods for nutrients present at low concentrations.
This is also a qualitative term. A method is applicable within the context in which it will be used, for example the analysis of a specific food matrix. Applicability relates to the freedom from interference from other constituents in the food or from the physical attributes of the food matrix that would make extraction of the analyte incomplete. Applicability is also determined by the usable range of the method. Methods that are applicable at high concentrations may not be applicable at low concentrations. Equally, a method may be applicable to one matrix (e.g. meat) but be inappropriate for another (e.g. a cereal product).
All unfamiliar methods or methods described for a specific food must be checked carefully when used for a matrix that is different from those for which it has been used previously.
Specificity is the ability of a method to respond exclusively to the substance for which the method is being used. Many methods are “semi-specific”, relying on the absence of interfering substances in the food being examined. Sometimes a method with poor specificity is acceptable when the purpose of the analysis is to measure all similar substances within a group (e.g. total fat, ash).
Accuracy is defined as the closeness of the value obtained by the method in relation to the “true value” for the concentration of the constituent. It is often expressed as percentage accuracy. Inaccuracy is, as a corollary, the difference between the measured value and the “true value”.
The concept of a “true value” is, of course, hypothetical because the “true value” for a nutrient in a food is not known. All analytical values are therefore estimates of that value.
Büttner et al. (1975) take the view that there exists a true value for all constituents in a sample of food. This is fundamental to the analysts' art; it is not true that the value for a defined analytical sample of a food is the “true value” for all samples of that food. The sampling error and the analytical errors for any specific method determine the confidence limits for all determined values.
The accuracy of a method is usually determined by reference to standard amounts of the analyte and preferably by the analysis of standard reference materials (SRMs) or certified reference materials (CRMs) that have been analysed, often using several compatible methods, by a group of skilled analysts to provide certified values together with the confidence limits of that value.
Precision is a measure of the closeness of replicated analyses of a nutrient in a sample of food. It is a quantitative measurement of “scatter” or analytical variability. Strictly speaking, it is imprecision that is measured by carrying out replicate analyses on the same sample (which must be homogeneous and stable). The measurements may be made by one analyst within one laboratory when the assessment is designated “repeatability” (that is, within-laboratory precision) or by several analysts in different laboratories when it is designated “reproducibility” (that is, between-laboratory precision). Comparisons can also be made among different analysts in one laboratory (called “concordance”), and by one analyst on different occasions.
In each case the standard deviation (SD) of the analytical values is calculated (which means that there must be a sufficient number of replications). The SD is customarily divided by the mean value to give a relative standard deviation (RSD), or multiplied by 100 to give the coefficient of variation (CV). In analytical literature, the RSD is used for reproducibility and rsd for repeatability.
It is important to recognize the distinction between accuracy (see the definition above) and precision. One can have very high precision (a low RSD) and poor accuracy and, conversely, have high accuracy with poor precision where the confidence limits of the value obtained will be wide. The ideal is to combine high precision (low RSD) with high accuracy (as judged by the value obtained with an SRM).
Detectability is defined as the minimum concentration of analyte that can be detected. This is rarely an issue in nutritional studies, as very low concentrations of nutrients, even some trace elements or vitamins, are not usually nutritionally significant. These are customarily recorded as “trace” in many printed food composition tables. However, it is useful to know whether or not a nutrient is present, and at what level one can confidently record zero in a database. The detectability limit of a method is the concentration at which the measurement is significantly different from the blank. Since blank values also show some variability, the limit can be defined as greater than +2SD (of the blank measurements) above the blank level. The detection limit is below the concentration at which measured values can be made; that is, it is outside the usable range of the method.
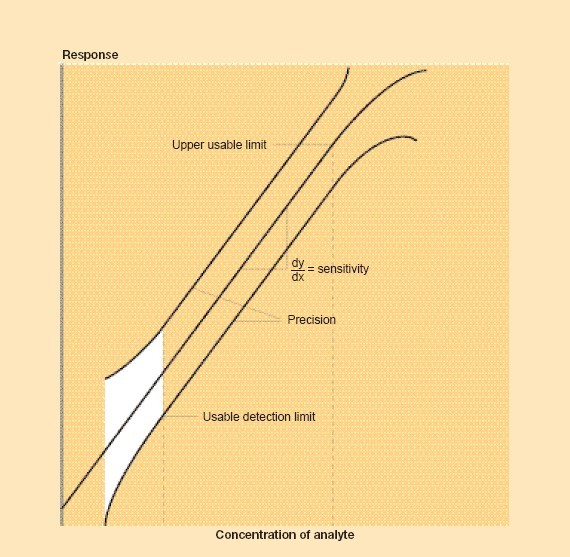
Sensitivity in analytical terms is the slope of the response–concentration curve or line (Figure 6.1). If the slope is steep the method has a high sensitivity; conversely, if the slope is shallow the method has a low sensitivity. When a narrow range of concentration is of interest, a high sensitivity is often desirable; for a wide range of concentrations, a low sensitivity may be preferable. In most nutritional composition studies, trace element analysis requires high sensitivity. In practice, this can often be achieved by increasing the response signal strength by electronic amplification or through chemical concentration of the element.
High sensitivity is usually required for the analysis of contaminants. While contaminants are not usually included in food composition databases, they may become more important in the future, especially those with antinutritional or toxicological properties.
Figure 6.1 Response as a function of concentration, illustrating the attributes of methods
Source: Modified and reproduced with permission from Stanley L. Inhorn, ed., Quality assurrance practices for health laboratories. Copyright 1978 by the American Public Health Association.
This is a qualitative attribute and refers to the capacity of a method to perform adequately in the face of fluctuations in the analytical protocol. Such fluctuations could include the timing of stages, changes in temperature, or the precise concentrations of reagents. It also includes variations in the skill, training and experience of the analysts carrying out the method.
Ideally, during the initial development of a method its authors should have explored and documented the capacity of the method to withstand these types of fluctuation and to perform under a variety of conditions. Methods are available for examining such variations (Youden and Steiner, 1975).
Authors of analytical methods should identify the stages in their methods that require strict attention and control, and document these in the published description of the method.
Figure 6.1 provides a diagrammatic summary of the attributes. In the figure the response (height, area, weight, volume, time, optical density or another type of measurement) is shown as primarily a linear function up to a certain level that defines the usable range of the method. Where only a single analyte elicits the response, the method is specific; this specificity may be inherent in the method or may be achieved by chemical separation from interfering substances. This, therefore, is a property of the chemistry of the analyte and of potential interfering substances. The sensitivity of the method is indicated by the slope of the response line. The confidence envelope indicates the precision of the method and the difference between the response line and the hypothetical true line represents the measure of accuracy. The confidence envelope can be calculated at any level, but 95 and 99 percent are commonly used. In the former case, only 1 in 20 measurements can be expected to fall outside the envelope and in the latter only 1 in 100. The white area represents the region of uncertainty where the relative standard deviation is so large that no certainty can be assigned to a value.
Even well-established methods need to be evaluated by the analysts themselves, using their own staff, reagents and equipment (Wills, Balmer and Greenfield, 1980). An evaluation of the attributes of the method should be established under the conditions prevailing in the laboratory and the performance characteristics that are relevant to the purpose of the analyses should be quantified.
In the first stage of the evaluation, the analysts should familiarize themselves with the method as described in the formal protocol for the method concerned. This begins with a “paper exercise” to ensure that the principle of the method is understood and that the various stages are clear in the analysts' minds. The list of reagents required should be checked against the procedures. Occasionally, a common reagent will be omitted from the reagent list because the authors assume that all laboratories will have it to hand. Standardization of some reagents may be needed before the method is started. At the same time, the analysts should check the equipment required and any specifications listed for the equipment.
Finally, the analysts should go through each stage, familiarizing themselves fully with its purpose. At this point it is suggested that an assessment of the criticality of each stage is made, as recommended in the ANALOP approach (Southgate, 1995); this exercise will determine the possibility for error or uncertainty that might occur if the conditions described are not followed precisely.
Timing may or may not be critical. For example “leaving overnight” may imply a specific time period, say from 18.00 to 09.00 the following day (i.e. 15 hours), or merely that when this point is reached the method can be left until the following day – an indeterminate time period. Timing may represent a minimum time period; alternatively, “heat for 10 minutes in a boiling water-bath”, for example, may mean “exactly 10 minutes” or “while the analyst takes coffee”. Understanding the critical timed stages is especially important when a method is carried out for the first time and until it becomes “routine”.
Analogously, the concentrations of certain reagents are also critical, especially when the reagent must be used in excess for a reaction to be fully completed.
Using the published description of a method as one would follow a recipe in cooking can be fraught with disaster. The analyst must understand the logic of a method. Running through a method as a trial and discounting the results is useful for checking the stages, especially with regard to timing. Less-experienced staff may take time to adjust themselves to a procedure for which the published account of the method suggests that there are many critical operations (e.g. as in the non-starch polysaccharide method [Englyst, Quigley and Hudson, 1994], where the mixing stages are critical). Once this assessment is completed, the analyst will be in a better position to evaluate the various performance attributes.
The application of an unfamiliar method to a food matrix other than that for which it was developed or used previously requires careful consideration. It will be necessary to decide, often intuitively, how the matrix will behave in an extraction phase and whether there is any likelihood of interfering substances being present. The chemistry of the analyte and the expected range of the nutrient in the “new” food will therefore need to be considered.
Such matters cannot always be decided intuitively, however, and the method must be tested on the food material. The use of different analytical portions will provide evidence of interference or indicate possible problems with extraction or inadequate concentrations of reagents.
The recovery of standard amounts of the analyte added to the sample can establish whether extraction is complete. Recovery tests are not completely adequate because the added analyte may be more easily extractable than the intrinsic nutrient. Poor recoveries indicate problems; good recoveries may be regarded as encouraging but not conclusive.
Comparisons with values reported in the literature for the matrix may be helpful, as may collaborative studies with another laboratory.
Assessing this attribute requires knowledge of the chemistry of the analyte and the food matrix. A value may be required for a group of substances, such as total fat (lipid solvent soluble) or sugars, in which case a semi-specific method may be adequate. Values for triacylglycerols or individual sugars, however, require a much more specific method. Certain vitamin values must include all the active forms; for example, vitamin A (retinol) values should include other active retinoids. Here again, specificity is critical.
This is a difficult attribute to measure because its true value is unknown. The first stage is to analyse standard amounts of the pure analyte. Recovery studies of standards added to the foods are useful, especially if a series of different amounts is used and then a comparison made of the sensitivity of the method for pure standards and the added standards. Recovery studies, as mentioned above, do not provide unequivocal proof of the accuracy of a method because they assume that the added nutrient may be extracted with the same efficiency as the intrinsic nutrient (Wolf, 1982).
Analysis of authentic samples that have already been analysed by another laboratory is a useful guide for analysts using a method for the first time. This procedure forms what might be regarded as a simple type of collaborative study.
Standard reference materials are unique materials with a range of food matrices (limited at present but increasing in numbers) that have been produced by a national or regional organization such as the National Institute of Standards and Technology (NIST, 2003a) in the United States or the Community Bureau of Reference (BCR) for the European Union (BCR, 1990; Wagstaffe, 1985, 1990). The samples have been very carefully homogenized and rigorously tested for homogeneity and stability under different storage conditions for different lengths of time (Wolf, 1993). They are then analysed using well-defined analytical methods. Where possible, a number of different compatible methods based on different principles are used. The values generated are then certified with defined confidence limits for the values. The range of nutrients for which SRMs or CRMs are available is limited (but increasing). Coverage is good for many constituents, including some trace elements, some fats, fatty acids, total nitrogen and cholesterol.
SRMs (or CRMs) are expensive to produce and therefore too costly to use routinely (say, with every batch of analyses – which would be the ideal). Each laboratory (or a group of local laboratories) should therefore consider preparing in-house reference materials using similar approaches to that used to produce SRMs (Southgate, 1995).
The homogenized material is stored in a large number of individual containers and used routinely in the application of the method and occasionally alongside the SRM. Recording the values obtained over time on a control chart will help identify any trends towards high or low values. A control chart usually has a central line indicating the control limits for a statistical measure (SD for example) for a series of analyses (American Society for Quality Control, 1973). The laboratory results are plotted on the vertical axis against time (days, hours, etc.) on the horizontal axis. The horizontal scale should provide for at least three months of data and the chart should be checked regularly for evidence of runs above or below the central line or any evidence of lack of randomness (Mandel and Nanni, 1978; Taylor, 1987). Theoretically, the values should be randomly distributed about the central line. When they fall consistently above (or below) the line, they represent possible indicators of systematic bias in the method, which should be investigated.
The preferred materials for in-house reference materials are non-segregating powders such as non-fat milk powders, gelatine, flours, powder mixes for parenteral feeds (Ekstrom et al., 1984) and food matrices common to the local food supply, e.g. soybean meal and fishmeal for ASEANFOODS (Puwastien, 2000). Torelm et al. (1990) describe the production of a fresh reference material based on a canned meat.
One alternative is to carry out analyses using standard samples on a routine basis using a control chart to alert laboratory personnel to problems requiring remedial action.
The original published description of a method usually gives some indication of the level of precision achieved in collaborative studies, thus providing a “standard of achievement”. Each laboratory, once its personnel are familiar with the method, should evaluate its own levels of precision.
The first step is for each analyst to assess their repeatability by analysing several replicates (preferably at least ten) of the same material and calculating the relative standard deviation. Second, all the analysts within the laboratory should analyse several replicates (preferably ten) of the same material to assess concordance within the laboratory. When setting up a method for the first time, it is useful to test repeatability and concordance using standards. Using blind concentrations of standards prepared by colleagues gives further confidence when using an unfamiliar method.
Finally, participation in a collaborative trial to assess the reproducibility of the method and to evaluate the laboratory repeatability with other analysts is a valuable approach that can be useful as part of the development of analytical skills.
Formal schemes exist for the collaborative analysis of some nutrients; samples for analysis are provided on a regular basis by NIST (2003a) in the United States and by the National Accreditation of Measurement and Sampling (NAMAS) in the United Kingdom (UKAS, 2003). In addition, Wageningen University in the Netherlands is the base for the International Plant-analytical Exchange (IPE, 2003), which provides a basis for developing analytical proficiency, especially for trace elements.
Difficulties may be encountered with regard to the entry of food materials into certain countries and most schemes are quite expensive, which may be a prohibiting factor where resources are limited. In such cases, the organization of local collaborative studies should be considered.
There are three major types of collaborative study. The first type, sometimes known as a “round robin”, or “ring test”, provides comparative assessments of laboratory performance. Homogeneous samples of food, often with their identities concealed, are distributed centrally, together with guidance on the preparation of standards and the calculation of results. The results are then collected centrally and analysed statistically. The results are usually provided to the participating laboratories in the form of charts showing the performance of each laboratory against the analyses as a whole. Each laboratory is given a code number and can assess its own performance. Outliers where the values obtained are significantly different from the mean and reproducibility found in the trial are also indicated. This type of collaborative study is of most benefit to laboratories involved in compositional analysis that wish to test and improve their performance.
A second type is that used by the Association of Analytical Communities (Thompson and Wood, 1993; AOAC International, 2003) to establish the performance of a method. In this case the collaborating analysts analyse a series of food samples supplied centrally, using a common analytical protocol. Standards and some reagents, where the specifications are critical (such as enzymes), are also supplied centrally, as are forms for calculating, expressing and recording the results. At least eight, but preferably more, analysts and laboratories are involved in such a study. The results are collected and analysed statistically, usually by an associate referee. The performance characteristics are used in the assessment of the method before it is accepted into the Official Methods manual.
A third type of study is used by the BCR in the European Union, primarily in the development of standard certified materials. Here, a group of laboratories analyses samples provided centrally, initially using their routine methods. Standards may be distributed together with forms describing how the results should be expressed. The results are collected centrally and analysed statistically. The findings are distributed and the analysts subsequently called to a meeting. The object of the meeting is to assess the different methods and identify where laboratories using the same methods found different values. Agreement is then reached on protocols that should be followed in a second round.
The results from the second round of the study will often identify methods that give satisfactory reproducibility and those methods that give similar results, although a third round may be required. These methods are then used in a carefully controlled certification study of food materials intended for potential reference materials. The ideal is to have a number of methods, based on different principles, that are compatible. In some instances the certification can only be given for values obtained by only one method.
It is important that the analysts involved in collaborative studies of this nature see the primary objectives of the studies as raising standards of analytical performance and furthering the development of analytical skills and not as a management tool for checking the performance of analysts.
When anomalous results appear in collaborative studies or in routine analyses, for example on the control charts, the first step is to go through the logic and application of the calculations, as these are the most frequent causes of anomalous results. Most collaborative studies define the calculations explicitly to avoid such problems, but they still occur. For this reason the calculation procedures should be set out in a logical fashion within the analytical protocols.
The second stage is to repeat the analyses with a series of freshly prepared standards. Improper dilutions or weighing are frequent causes of error.
In the third stage the analyses are repeated by another, more experienced, analyst. Repeating the analyses using a portion from an earlier stage of the analyses does not constitute a rigorous check; ideally, fresh analytical portions should be used. Neither does simple repetition provide an adequate check because any bias related to the standard or the food matrix may be replicated.
If the results still appear anomalous the analyst should analyse the sample blindly using only its sample code number and, if possible, a colleague should be asked to introduce a “blind” replicate. Southgate (1987) has identified a range of laboratory practices that may lead analysts to believe, erroneously, that they have achieved good repeatability and how these practices can be changed (Table 6.2).
All these operations form part of a data quality assurance scheme and their documentation is vital for database compilers when they come to assess the quality of the analytical data, which is discussed in Chapter 8.
Table 6.2 Operational practices that may lead to systematic errors | ||||
|
Operation |
Common practices |
Remedy |
||
Size of analytical portion |
Identical or closely similar analytical portions |
Work with replicates of different sizes |
||
Reagents used |
Always from same batch |
Vary sources of reagents |
||
Standard solutions |
Prepared from same stock or same series of dilutions |
Prepare fresh standards regularly |
||
| Replication of analyses |
Analysed in same batch or at the same time |
Analyse replicates in different batches or different days. Participate in collaborative studies |
||
| Analyst | Only one analyst | Carry out analysis with different analysts regularly. Collaborate with other analysts Exchange samples |
||
| Choice of procedure | Only one procedure | Where possible, use methods based on different principles. Collaborate with other laboratories |
||
Source: Modified from Southgate, 1987. | ||||
![]()
![]()
![]()