![]()
![]()
![]()
J.C. Hanekamp;1 R.J. Wijnands2
Introduction
Analysis technology, food safety related risk characterisation and legislation have collided globally over the issue of zero-tolerance regulation. The detection in 2001 of chloramphenicol (CAP) in shrimp imported into Europe from Asian countries triggered this confrontation.3 A number of other substances that are part of the so-called Annex IV list of the European Council Regulation 2377/90 (the 'MRL Regulation'; Maximum Residue Limit)4, for which a zero-tolerance regime is in place, also surfaced in food-products analysis and intensified the public, political and scientific debate. In this article we will give a precis of the issues at hand and will propose a coherent and logical approach of veterinary drug residue legislation based on the following universal points of reference:
Focussed on food safety as such in order to measurably guarantee public health
Contribute to a global economic and regulatory level playing field
Sensitive to innovation in the pharmaceutical and food and feed industry
Sensitive to corresponding authorisations in the human clinical environment
These four points of reference will be developed specifically for veterinary residues in food-products. Although there are both national as well as trade-zone and even global residue standards, our focus will be on the methodology of standardisation. As said, actual threshold values may vary.
|
Intermezzo I: CAP, risks and Annex IV5 The risk of CAP-related aplastic anaemia -the primary risk of CAP exposure- surfaced and was documented as a consequence of human therapeutic use. Combined with the fact that no ADI (Acceptable Daily Intake) could be established -for lack of adequate scientific data and scant evidence for carcinogenicity- CAP was placed on the Annex IV, whereby a veterinary ban was put in place. CAP, however, is still registered as a human therapeutic substance. Figure 1 CAP exposure level differences between therapy and food residues
CAP exposure through food never yielded any documented cases of aplastic anaemia. (Evidently, absence of proof does not infer proof of absence.) The incidence of aplastic anaemia as a result of therapeutic CAP exposure is estimated to be 1:10 000 000, which is a factor 10 below the MTR (Maximum Tolerable Risk being a 1:1 000 000 added mortality in the human population). In relation to CAP clinical treatment as such, 1 case of aplastic anaemia in approximately 30 000 or more therapy courses has been estimated.6 Ocular use of CAP has been weakly associated with aplastic anaemia and the risk is probably less than one in a million treatment courses.7 The cancer risk of CAP food-residue exposure is estimated to be at least 5 000 below the MTR.8 |
A précis of the zero-tolerance matter
The application of pharmacological active substances in the production of animal food-products usually will under certain conditions result in the presence of residues in the end product. As a rule this will not pose a quantifiable risk to consumers. Council Regulation 2377/90 provides for three general legal tools to address the potential toxicity of residues of veterinary medicinal products:
1. Establishment of MRLs for a substance (Maximum Residue Limits, Annex I and III)
2. Approval of a substance without the establishment of MRLs (Annex II)
3. Prohibition of the use of a substance (Annex IV)
Prohibition of a substance -by inclusion in Annex IV- is a result of the impossibility to establish an Acceptable Daily Intake (ADI) and therefore a MRL. However, risks associated with these substances usually surface visibly as a result of human therapeutic use -such as in the case of CAP- or are extrapolated from experimental data. These substances are subsequently regarded as hazardous to consumers irrespective of exposure-levels. This classification by inference results in the prohibition of the use of the substances in veterinary medicinal products, despite the fact that the products might be fully authorised as a human clinical substances (CAP, furazolidone. metronidazole, and the like).
Zero-tolerance is intended to eliminate certain presumed risks to human health as a result of exposure to residues in animal food products. This in effect means three things: (i) detection as such, irrespective of concentrations, is deemed a public health risk (displaying the regulatory choice for the Linear Non-Threshold model (LNT) from which the risks are inferred); (ii) detection in food is singularly related to the illicit veterinary use (intended misuse) in food production; (iii) a zero-tolerance approach in food safety of illicit veterinary use would remove the residue (and its concomitant risks) from the food chain and its products.
|
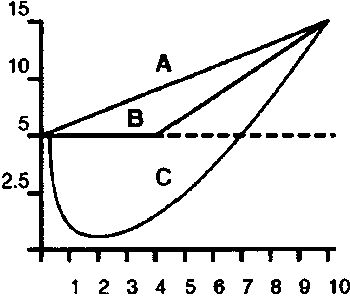
Intermezzo II: Toxicological models9 There is solid scientific evidence that the extrapolation from high-level exposures -of which detrimental effects can be observed- to low-level exposures -of which potential detrimental effects cannot be observed-, in general is not linear. In the absence of knowing by what mechanism of action a toxicant may harm individuals, regulatory toxicology assumes that even tiny doses can cause injury. Roughly two models to determine the dose-response relationship have traditionally been used in toxicology in the assessment and regulation of risks of toxicants: the threshold model (B) is used in the assessment of risks for non-carcinogens, and the Linear Non-Threshold (LNT) model (A) to extrapolate risks to very low doses of (genotoxic) carcinogens. The risks associated with taw-level exposures of Annex IV substances are singularly derived from the LNT axiom. Figure 2 Three toxicological extrapolating models
[In the above figure tumours per animal are depicted on the y-axis, with the related dose on the x-axis. The animal control group (not exposed to the carcinogen) is depicted by means of the black horizontal dotted line at the 5-level on the y-axis. The hormetic model C predicts a lower amount of tumours than the control group when exposure levels of the carcinogen are below 7. The hormesis concept challenges the axiom and use of low-dose linearity in estimating cancer risks, and emphasizes that there are thresholds for carcinogens.] However, it has been contended persuasively that the most fundamental shape of the dose-response is neither threshold nor linear, but U-shaped (C), and hence both current models provide less reliable estimates of low-dose risk. This U-shape is usually referred to as hormesis: a moderate stimulation of response at low doses and an inhibitory response at higher doses. It is to be regarded as an adaptive response of an organism towards toxicological perturbations. Acceptance of hormesis suggests that low doses of toxic/carcinogenic agents may reduce the incidence of adverse effects. Hormesis redefines the concept of 'pollution' and 'contamination'. It questions the premise that 'pollutants' are unreservedly bad. This is revolutionary because modern environmental and public health legislation is built in large part on the moral dichotomies of good versus evil, clean versus dirty, natural versus unnatural. |
Flaws
The fatal flaw of zero-tolerance is that 'zero concentration' -implied by zero-tolerance- is not a physico-chemical reality as it contradicts the Second Law of Thermodynamics. (One of the implications of the Second Law is that complete separation of for instance a binary mixture (a mixture of two components) is not possible.) This mere fact leaves the Annex IV without foundation. Indeed, increasing sensitivity of analytical equipage will reveal -now or in the future- ever-smaller amounts up to the molecular level of listed substances, whatever the source. Residue regulation based on the MRPL (Minimum Required Performance Limit; the concentration level that regulatory laboratories in the European Community should at least be able to detect and confirm) will therefore eventually fail, as it will be prone to technological development as shown below for the history of limits of detection of CAP.10 More importantly. MRPL-based regulation is devoid of any toxicological deliberations and is therefore in terms of protection of public health irrelevant. Below an example is given of decreasing limits of detection:
Figure 3 Improvement of limits of detection of CAP in
milkpowder (Instrumental methods are denoted by
 and the solid line; screening
methods by
and the solid line; screening
methods by  and the broken
line.)
and the broken
line.)
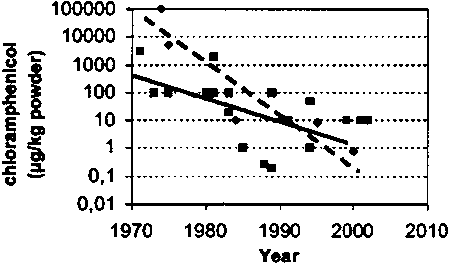
Additionally, zero-tolerance as defined by the Annex IV has the explicit goal of zero-risk. In view of recent jurisprudence this is unlawful.11 Proof of absence ('zero-concentration'), and thereby proof of no harm constitutes a probatio diaholica.
|
Intermezzo III: Multiple Sources of semicarbazide and CAP12 SEM (semicarbazide) has long been considered a characteristic molecule of the antibiotic nitrofurazone (Annex IV). Studies have shown that the parent drugs are rapidly metabolised by animals, and are therefore undetectable directly. The stable metabolites are however detectable for some weeks after application of nitrofurans and are therefore regarded as indicators for the application of nitrofurans. However, recently SEM was found as a contaminant in food packaged in glass jars, unrelated to the nitrofurans. In this case SEM is formed by thermal degradation of azodicarbonamide (ADC). ADC is used as the blowing agent in plastic gaskets of packaging material. SEM migrates from the gaskets into food products. SEM was also detected in special animal and vegetable matrices that had been concentrated using drying procedures like heating to reduce water content. A substantial formation of SEM was observed after samples were treated with hypochlorite (bleach) in accordance with common food processing methods used for disinfection or bleaching.13 CAP has unequivocally been found in German sewage and surface water.14 The most likely source for CAP in the aquatic environment in this case is human medicinal use. Other research has shown that in food products not related to illicit use, CAP can nevertheless be detected (CAP is a naturally produced antibiotic). These results are suggestive for multiple sources of CAP in the food-production chain, although definitive proof is lacking. However, the fact that in Europe CAP can actually be detected in the aquatic environment does give rise to the distinct possibility that the food-production chain can be contaminated through other routes than intended misuse. Moreover, the aquatic environment (including groundwater) proves to be a source for multiple antibiotics by which the public is exposed through mainly drinkingwater.15 |
Zero-tolerance intrinsically demands a continued development of analytical equipage, so that limits of detection will continue to drop. With the increased capabilities of analysis the chances of detection of banned substances will by definition increase (see intermezzo III): modem analysis has spawned the 'vanishing zero'. This will indubitably lead to the destruction of increasing amounts of food and augment inequalities between countries with different levels of scientific sophistication. Zero-tolerance as part of food safety therefore can and will be misused as a trade-barrier. More importantly, the regulatory barrier between banned veterinary and authorised human therapeutic products will predictably be traversed through the aquatic environment. CAP could well be the first example of this quandary.16
Tools of Innovation
Food safety is the highest priority of the (inter)national regulatory bodies and the international producing and trading industries. Tools of regulatory innovation need to focus on food safety as such. The following regulatory tools of innovation could serve this purpose:
Risk assessment and management
MTR (Maximum Tolerable Risk level)
Toxicologically Insignificant Exposure levels (TIE)
An 'Annex IV' based on proof of harm of low-level exposure toxicity
Internationally harmonised veterinary products authorisation en lieu with authorised human medicinal products
(Partial) integration of risk assessment, management and communication
The abovementioned list of criteria expounds Normal Intended Use. By that we mean that clinical substances applied in the animal-producing field are authorised products used with a normal intentional purpose. This means that food-safety regulation is not meant to tackle intended misuse. Again, its focus is on safeguarding public health. Below we have depicted a decision tree by which future regulation could best be organised:
Figure 4 Food safety regulatory decision tree for clinical residues
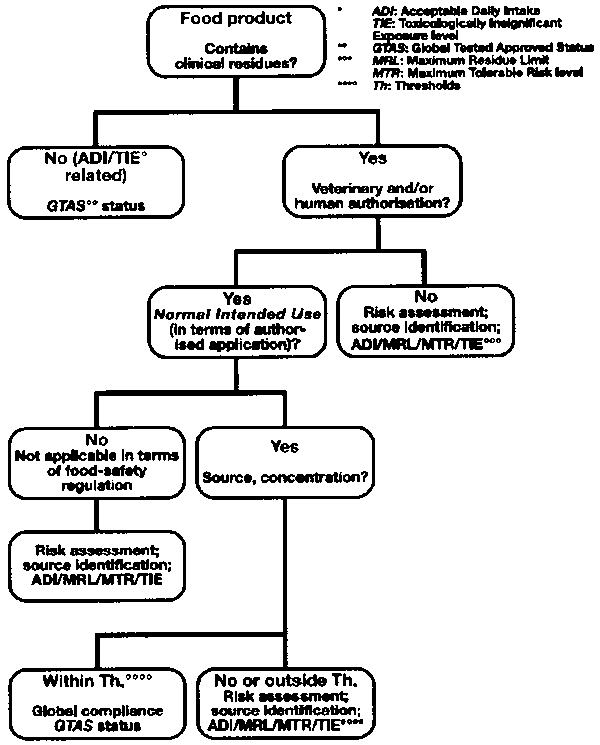
The buttress of the decision tree is the risk approach, as 'zero-concentration' is not a physio-chemical reality. A number of risk tools are on hand namely, the Maximum Tolerable Risk level (MTR), the Acceptable Daily Intake (ADI; and the related Maximum Residue Limit (MRL)) and the Toxicologically Insignificant Exposure level (TIE). We propose a TIE for banned substances as to preclude analytical progress as the sole limiting factor for the determination of regulatory compliance. Research of indirect additives in food, based on the Carcinogenic Potency Project,17 suggests a TIE of 0.5 ppb. l8 This TIE level is all the more pertinent in view on the current scientific dialogue on hormesis (see Intermezzo II).
Pharmaceutical innovation requires responsive legislation, which gives clear guidelines concerning authorisation and compliance. The MTR serves best as a risk assessment and management criterion as it is internationally accepted and recognised (MTR; being a 1:1 000 000 added mortality (morbidity) risk in the human population).
As shown in the above depicted decision tree, normal intended use is not limited to authorised veterinary use but also includes human clinical use as the aquatic environment can potentially be a source of clinical residues for the food-production chain. Obviously, concentrations present in food as a result of exposure of clinical products through the aquatic environment are much lower than normal intended veterinary use. (Drinkingwater derived from either surface- or groundwater contains numerous excreted clinical compounds at very low levels not associated with any measurable risk for human health, whereby a strict risk-avoiding approach in terms of zero-tolerance within the food sector is disproportionate.) The inclusion of the normal intended use of human medication as a result of which the food chain is exposed to these substances (through the aquatic environment) is a logical consequence of the risk-based approach proposed here and resolutely widens the view-screen of risk-based food safety regulation. Therefore, the relationship between human and veterinary clinical substances could best be depicted as follows when risk assessment strategies are used as a central theorem:
Figure 5 Relationship between human and veterinary clinical substances

In order to preclude future regulation leading to an unlawful probatio diabolica, it is essential that proof-of-harm of low-dose toxicity supersede the current Annex IV regulation. (When proof-of-harm as a result of food residue exposure surfaces, then the substance needs to be listed on an amended 'Annex IV.) Lack of data to establish a MRL as such is not a sufficient ground to ban certain veterinary products, even more so when those products are authorised as human medication and damaging effects surface only as a result of human therapeutic use. Indeed, with any authorised human and veterinary medication a balance is struck between toxicity and beneficial effects at the biological active dosage. Risks materialising at the human therapeutic level are not indicative for a veterinary ban (see intermezzo I and II).
The risk approach should have an international jurisdiction in order to avoid trade barrier issues based on analysis non-compliance. Therefore a Global Tested Approved Status is introduced in the decision tree. Tools of analysis need to be horizontally unified in order to generate global compliance and a level playing field as to preclude trade-barriers. Trade between nations will benefit from international cross-compliance, in which properly analysed goods will be accepted unreservedly by importing nations. In cases where non-authorised substances are detected, a risk analysis of observed concentrations needs to be undertaken with food safety as the sole objective.
References and notes
1 To whom correspondence should be addressed: [email protected]; +31(0)793460304.
2 Dr. R.J. Wijnands is director of the Dutch Confederation of Buildingmaterial Producers.
3 J.C. Hanekamp, G. Frapporti and K. Olieman. Chloramphenicol. food safely and precautionary thinking in Europe. Environmental Liability. 2003, 6, 209 - 221.
4 Council Regulation (EEC) No. 2377/90 of 26 June 1990 laying down a Community procedure to set up maximum residue limits of veterinary medicinal products in foodstuffs of animal origin, Official Journal L224 18 August 1990, 1 - 8.
5 See note 3.
6 IPCS-INCHEM (Chemical Safety Information from Intergovernmental Organizations), webpage http://www.inchem.org/documents/jecfa/jecmono/v33je03.htm (last visited on the 5th of July 2004).
7 J.R. Laporte, X. Vidal, E. Ballarin. L. Ibanez, Possible association between ocular chloramphenicol and aplastic anaemia-the absolute risk is very low. British Journal of Clinical Pharmacology, 1998, 46(2), 181-184.
8 P.A.H. Janssen. A.J. Baars and M.N. Pieters. Advies met betrekking tot chlooramfenicol in garnalen. 2001. RIVM/CSR. Bilthoven, The Netherlands. [Recommendations on chloramphenicol in shrimp]
9 E.J. Calabrese and LA. Baldwin, Toxicology Rethinks its Central Belief. Hormesis Demands a Repraisal of the Way Risks are Assessed. Nature, 2003. 421, 691 - 692.
E.J. Calabrese and L.A. Baldwin, Hormesis: the dose-response revolution. Annual Review of Pharmacology and Toxicology, 2003. 43, 175-197.
K.K. Rozman and J. Doul, Scientific foundations of hormesis. Part 2. Maturation, strengths, limitations, and possible applications in toxicology, pharmacology, and epidemiology. Critical Reviews in Toxicology. 2003, 33 (3-4), 451 - 462.
F.B. Cross, Legal Implications of Hormesis. Humans & Experimental Toxicology, 2001, 20(3), 156- 158 (see also http://www.belleonline.com/n2v92.html last visited on the 5th of July 2004).
J.B. Wiener. Hormesis and the Radical Moderation of Law. Human & Experimental Toxicology, 2001. 20(3). 162 - 164. (see also http://www.belleonline.com/n13v92.html last visited on the 5th of July 2004).
BELLE (Biological Effects of Low Level Exposure) www.belleonline.com (last visited on the 5th of July 2004).
See also on toxicological models: C.R. Sunstein, Risk and Reason. Safety, Law and the Environment. 2002, Cambridge: Cambridge University Press.
10 See note 3.
11 Policies aimed at the exclusion of risk or that generate an impossible burden on economic parties in terms of proof of no- harm is regarded as unlawful as is discussed in a verdict by the Court of First Instance (Case T13/99 Pfizer Animal Health SA. 130):
'130. Supported more specifically by Fedesa and Fefana, Pfizer submits that in any such risk assessment, the Community institutions must show that the risk, although it has not actually become a reality, is nevertheless probable. The existence of a 'very remote risk' should be allowed given the concrete positive elements arising from the use of the product concerned. In any event, the Community institutions cannot legitimately apply a test which Pfizer describes as a 'zero risk test. Such a test is inappropriate since it is impossible to satisfy. It amounts essentially to requiring probatio diabolica from the industry, something which is recognised as unlawful in all the legal systems of the Member States (Opinion of Advocate General Mischo in the Greenpeace case cited at paragraph 115 above, ECR I-1651, at I-1653, point 72). It is never possible to prove conclusively that a chemical or pharmaceutical compound or anything created by modern technology represents a zero risk to public health now or that it will do so in the future. To apply such a test would quickly lead to the paralysis of technological development and innovation.'
12 See note 3.
J.C. Hanekamp. J. Kwakman, Beyond zero tolerance: a new approach to food safety and residues of pharmacological active substances in foodstuffs of animal origin. Environmental Liability. 2004, 1, 33 - 39.
13 See note 3.
14 R. Hirsch, T. Ternes, K. Haberer and K.-L. Kratz, Occurrence of antibiotics in the aquatic environment. The Science of the Total Environment, 1999, 22.5. 109 - 118.
15 K. Kummerer (ed.). Pharmaceuticals in the Environment. Sources, Fate, Effect and Risks. 2001, Berlin: Springer Verlag. R. Hirsch. T. Ternes, K. Haberer and K.-L. Kratz. Occurrence of antibiotics in the aquatic environment. The Science of the Total Environment, 1999, 225. 109 - 118.
J.F.M. Versteegh, A.A.M. Stolker, W. Niesing, J.J.A. Muller, Geneesmiddelen in drinkwater en drinkwaierbronnen. Resultaten van het meetprogramma 2002. 2003. rapport 703719004/2003. RIVM, Bilthoven, The Netherlands. [Medication in drinkingwater and drinkingwater wells. Results of the analysis series 2002.]
16 See note 3.
17 B.N. Ames and L.S. Gold. Paracelsus to parascience: the environmental cancer distraction. Mutation Research. 2000, 447, 3- 13.
See the Carcinogenic Potency Project at http://potency.berkeley.edu/.
See also L.S. Gold, T.H. Slone, N.B. Manley and B.N. Ames, Misconceptions about the Causes of Cancer (Vancouver, The Fraser Institute, 2002); available at http://potency.berkeley.edu/text/Gold_Misconceptions.pdf.
18 A.M. Rulis. Threshold of regulation: Options for handling minimal risk situations (1992), in: Food Safety Assessment, ACS Symposium Series 484. 132-139.
![]()
![]()
![]()