![]()
![]()
![]()
Each year in the United States millions of illnesses and thousands of deaths are traced to contaminated food, with an estimated cost from US$5 billion to over US$22 billion. Experts believe that the risk of food-borne diseases has increased during the last 20 years (United States General Accounting Office, 1996a). Food-borne diseases can originate from consumption of viruses or bacterial pathogens, toxic substances and parasites. It has been estimated that about half of all food-borne disease outbreaks remain unrecognized, primarily due to inadequate diagnostic methods and sampling (Svensson, 2000).
Fish are implicated in 25 percent of food-borne disease outbreaks in the United States; 86 percent of them due to biotoxins, mostly ciguatera (Olsen et al., 2000; Valdimarsson, Cormier and Ababouch, 2003). The effect of these biotoxins, their chemical structures and methods of detection have been the subject of several reviews (Botana et al., 2002; Daranas, Norte and Fernández, 2001; Lehane and Lewis, 2000; Todd, 1993; WHO, 1984) and the reader should refer to those publications for detailed information. Table 1 gives some data on the most common toxins.
The classical method to detect toxins is the mouse bioassay: mice are injected with an extract of a suspect sample and their mortality is registered. The method is expensive, not-specific, highly variable, with low sensitivity, only a limited number of samples can be processed, it is slow, it requires specialized facilities and it involves the use of live animals. For all these reasons it is highly desirable to find a more humane, easier and cheaper method. The newer detection procedures focus on: i) detecting the toxic molecules themselves, using chemical methods; ii) detecting some part of the molecules by immunological methods; or iii) detecting the activity of the toxins, called bioassays. Lehane and Lewis (2000) give a review of different methods used to identify ciguatera toxins.
Usually, methods that aim at identifying the toxins themselves are not very user-friendly since they require specialized lab equipment and personnel for separation and detection; for example, chromatography for separation of toxins from the rest of the molecules in the crude extracts and fluorescence for detection (Franco and Fernández-Vila, 1993; Lawrence et al., 1991).
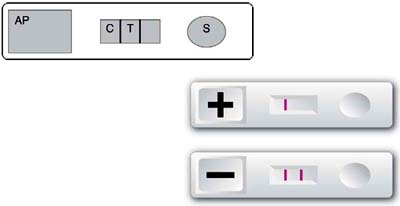
Immunological methods are usually easy to use and render fast results. Monoclonal antibodies have been used to develop methods to identify, among others, ciguatera toxins (Hokama et al., 1985, 1998), PSP and ASP toxins. Figure 1 illustrates the Jellet Rapid Test kit for the detection of PSP toxins which takes about 20 minutes (Jellet et al., 2002; Laycock et al., 2000). A similar kit has been developed for ASP and others are under development for other toxins. The kit is considered to be user-friendly and good for in situ and preliminary screenings (Jellet et al., 2002, Overrein, Asp and Aune, 2002). The most common problems with these types of techniques are that the part of the toxin molecule that elicits the reaction with the antibody may be altered in the sample (for example, due to cooking) with corresponding variation in the response (it may be a weaker positive); there may also be differences in the chemical structure of toxic molecules from different geographical locations, and the antibodies may fail to give comparable reactions with all of them (Jellet et al., 2002, Overrein, Asp and Aune, 2002).
|
FIGURE 1
|
The third type of analysis is called bioassay because it aims to identify the toxins by detecting their effects. Louzao et al., (2001, 2003) have described a method to detect PSP toxins which measures changes in the membrane potential of excitable cells, which are detected by a fluorimetric method. Basically, excitable (neuroblastoma) cells are incubated with a fluorescent dye, whose distribution across the membrane is potential-dependent, and with the sample under examination. If the sample contains PSP toxins, the toxin opens the Na+-channels of the cellular membrane, depolarizing and inducing a change in the membrane potential and a consequent change in the measured fluorescence (Louzao et al., 2001). The method has been later modified to be used on a microtitre plate, which makes it more user-friendly and susceptible to automation (Louzao et al., 2003). A similar method, also using neuroblastoma cells has been developed to detect ciguatera toxin (Manger et al., 1995), but in this case the membrane depolarization is detected by change in color in the medium: neuroblastoma cells are incubated with a tetrazolium compound (MTT) which is reduced by healthy cells to a blue formazan product. No change in colour is registered if MTT is incubated with dead cells: when the neuroblastoma cells are incubated with samples containing ciguatera toxin, the toxin binds to the Na+ channel receptors on the cells, thus damaging the cells and preventing them from reducing the MTT and therefore no colour change is noticed.
The advantage of bioassay-based methods is that since they detect toxic activities rather than molecules or parts of molecules they can better predict the harmful effects of a contaminated sample (regardless of whether the “correct” toxin is implicated in the toxicity) (Spaldin, 1995) and can easily be automated to analyse hundreds or thousand of samples per day (Louzao et al., 2003). Some disadvantages are that they are more sophisticated and expensive than immunological methods, they do require viable cells, and references therein. and therefore are probably not going to be suitable to be used in situ (for example at coastal locations suspected to be contaminated), and that only the effect on those cells used in the bioassay is measured. For the detection methods illustrated here this implies that effects other than membrane depolarization (for example hyperphoshorylation) will not be registered, although they may also be fatal in the short or long term. One would need a battery of bioassays (as one would need a battery of antibodies) to cover all possible poisoning mechanisms.
TABLE 1
Some biotoxins from toxic microalgae
|
Poisoning |
Toxins |
Mechanisms of action |
|
Paralytic shellfish poisoning (PSP) |
Saxitonins (SRXs) |
Life threatening neurotoxic agents: bind strongly to sodium channels and block them. |
|
Ciguatera fish poisoning (CFP) |
|
|
|
Group 1: lipid soluble toxins. |
Ciguatoxins (CTXs) and Gambierol |
Neurotoxic potent agents. Bind to sodium channels and open them. |
|
Group 2: water soluble toxins |
Maitotoxin (MTX) and |
Cause gastrointestinal, neurological and cardiovascular problems. Activates voltage-independent Ca++ channels, increasing intracellular Ca++ levels and all the subsequent cascade events. |
| |
Palytoxin |
Multiple: membrane depolarization, stimulation of arachidonic acid release, stimulation of neurotransmitter release, inhibition of Na+/K+-ATPase, induction of contraction of smooth muscle and tumor promoting activity. |
|
Diarrheic shellfish poisoning (DSP) |
Okadic acid (OA); Dinophysistoxins (DTXs) |
Potent inhibitors of protein phosphatases type PP1 and PP2A, causing protein hyperphosphorylation and tumorigenesis. |
| |
Yessotoxins (YTX) |
Does not inhibit phosphatases. Not induction of tumors. Assumed that health risks are less than those of other DSP toxins |
| |
Pectenotoxins (PTX) |
Not tumor promoters. Mechanism unknown. Hepatotoxic. |
|
Neurotoxic shellfish poisoning (NSP) |
Brevetoxins (BTXs) |
Similar to ciguatera, but less severe. Predominating gastrointestinal and neurological symptoms. Act on the 5 site of voltage sensitive Na+-channels. |
|
Amnesic shellfish poisoning (ASP) |
Domoic acid (DA) and its congeners |
Gastroenteritis. Acts as agonist to glutamate receptor, which conducts Na+ channels inducing depolarization which in turns increases Ca++ permeability and ultimately leads to cell death. |
|
Azaspiracid poisoning (AZP) |
Azaspiracid and its congeners |
Necrosis in the lamina propria of the small intestine and lymphoid tissues |
|
Other |
Amphidinols |
Antifungal, hemolytic |
|
Prorocentrolides |
Fast acting (in killing mice). Mechanism unknown. Not protein phosphatase inhibitors |
|
|
Pinnatoxins |
Ca++-channel activators |
|
|
Spirolides |
Not fully identified. Affect Ca++-channels |
|
|
Gymnodimines |
Similar to NSP toxins. Pharmacological action not identified |
Data from Daranas et al, [ 2001]
Ingestion of pathogenic viruses can cause polio, gastro-enteritis and hepatitis (see reviews by Lees, 2000; Svensson, 2000 and Table 2). Man is the only known reservoir for the major viruses causing food-borne diseases: calicivirus and hepatitis A virus (Svensson, 2000). As far as it is known, all the viruses that cause food-borne illnesses are transmitted via the faecal-oral route. Shellfish-borne gastro-enteritis was linked to viruses for the first time in the United Kingdom in 1976-77 when cooked cockles were epidemiologically linked to 33 incidents affecting almost 800 persons (Appleton and Pereira, 1977). A large outbreak involved over 2000 persons in Australia (Murphy et al., 1979), and other outbreaks have been registered in Japan, Canada, the United Kingdom and the Scandinavian countries (Lees, 2000).
The high relevance of viruses in food-borne diseases was not suspected until the sensitive molecular detection techniques became available in the 1990s. Caliciviruses, such as Norwalk virus, were then recognized as the major cause of seafood-associated gastro-enteritis: over 80 percent of the outbreaks of non-bacterial gastro-enteritis in the United States and Europe are currently attributed to caliciviruses (Svensson, 2000).
TABLE 2
Classification of viruses associated to food-borne human diseases. Data taken from Lees, 2000. Genogroup in Caliciviruses refers to the molecular classification based on nucleotide sequence homology of the capsid protein (Svensson, 2000)
|
Name |
Genome |
Morphology |
Types-(Genogroup) |
Recent classification |
Detection |
Novel |
Problems |
Relationship to seafood consumption |
|
Caliciviruses |
Positive sense single stranded RNA |
Round, 30-35 nm diameter, with cup shaped depression (“Star of David” morphology) |
Norwalk virus (I), Southampton virus (I), Hawaii agent (II), Norwalk like viruses (NLV: Snow Mountain agent) (II), Others |
Genus 1: NLV Genus 2: Sapporo like viruses |
Difficult to work with cannot be cultured by conventional virological techniques great progress by using molecular techniques NLV and Sapporo virus cloned and sequenced [Liu et al., 1995] |
Based on molecular techniques [Berke et al., 997; Clarke and Lambden, 1997] |
NLV are the major cause of epidemic gastrointestinal illnesses in families or community outbreaks. Infection occurs in all age groups and they are the major cause of gastroenteritis in adults [Kapikian, 1996; Kaplan et al., 1982]. Sapporo like viruses Genetically distinct from NLV. They may comprise several strains. |
NLV: Frequently associated to seafood (shellfish and bivalve) consumption Sapporo like viruses: Not documented to have caused diseases following seafood consumption |
|
Hepatitis E virus |
- |
- |
There may be one single serotype [Tsarev et al., 1999] |
Formerly a calicivirus, now not assigned to any group [Pringle, 1998]. |
Cloned and sequenced [Bradley, 1995] |
|
Symptoms similar to hepatitis A. Endemic in many developing countries, particularly in Asia. High mortality rate in pregnantwomen. |
Outbreaks mostly linked to consumption of contaminated drinking water. [Lees, 2000] |
|
Astroviruses |
Positive sense single stranded non segmented RNA |
Round, about 28 nm in diameter. Some particles display a star like shape |
Seven serotypes |
|
Difficult to grow in culture. Recently specialized cell lines have been used to grow astroviruses directly from stool samples [Wilcocks et al., 1990] |
|
Symptoms similar to those of NLV: vomiting, diarrhoea, fever and abdominal pain, mainly in children and elderly. Usually full recovery and immunity after natural infection. Occur throughout the world, mainly intemperate climates and during the winter. |
Their importance as causative agents of gastroenteritis following seafood consumption is unclear. Molecular techniques are expected to clarify this point. |
|
Rotaviruses |
11 separate strands of double stranded RNA |
Spherical viruses with cubic symmetry, about 72 nm in diameter and wheel-like morphology |
PAGE of the 11 strands gives information on virus strains. Serologically unrelated groups A to G. Most human isolates belong to group A, which comprises at least 7 serotypes |
|
Adapted to grow in cell cultures. Together with the large amounts of particles recuperated in stools has led to the development of diagnostic kits and assays [Yolken Wilde, 1994] |
Triplex reverse transcriptase PCR[Tsai et al., 1994]. |
Group A rotaviruses have been frequently identified as the most common viral pathogen in diarrhoea requiring treatment or hospitalization in children under 2. In developing countries they are considered to account for about 20% of all diarrhoeal associated deaths in children under 5. Not considered to cause significant problems in adults. Cause of Group A rotavirus infections are detectable with antibodies in virtually all children by the age of 5 |
The infection is common and the viruses are shed in large numbers in stools(>1012 particles/g faeces) leading to detectable levels in sewage waters. Rotaviruses have been detected in bivalve shellfish grown in contaminated waters but they have not been linked to infectious disease, may be due to resistance to severe infection developed by active immunity acquired by repeated infection throughout life [Bishop, 1996] |
|
Adenoviruses |
Double stranded non segmented DNA |
Symmetrical no enveloped icosahedron, about 80 nm diameter |
Six main subgenera |
|
Most are easily cultivable except enteric adenoviruses serotypes 40 and 41 |
PCR [Girones et al., 1995, Pina et al., 1998] Nested PCR [Vantarakis & Papapetropoulou, 1998] |
Associated with gastroenteritis [Wadell et al., 1994]. Reported to cause about 10% of infantile gastroenteritis(second only to rotaviruses). Less severe but longer than rotaviruses diseases. Shed into the gutand isolatable from faeces. |
Detected in polluted sewage effluents, seawater and shellfish [Girones et al., 1995, Vantarakis & Papapetropoulou, 1998] but no seafood related outbreaks have been, reported |
|
Enteroviruses (fam. Picornaviridae) |
Positive sense single stranded non segmented RNA |
Smooth, round non enveloped, about 27 nm in diameter. |
66 immunologically distinct serotypes are known to cause infections in humans, including polioviruses, coxsackieviruses (groups A and B), echoviruses and enteroviruses serotypes 68 to 71 [Muir et al., 1998] |
|
|
Nested PCR [Vantarakis & Papapetropoulou, 1998] Triplex reverse transcriptase PCR |
All groups may become infected. Often mild or unapparent. In some cases the virus can spread to organs other than the intestinal tract and cause serious or fatal diseases(f.e. poliomyelitis, acute myocarditis, aseptic meningitis, hemorrhagic conjunctivitis, congenital infection of neonates and other non-specific febrile illnesses). Concern that they may cause or contribute to chronic diseases such as diabetes miellitus. |
Isolated from sewage effluents and bivalve shellfish. Bivalve, shellfish have not been linked to transmission of enterovirus diseases. |
|
Hepatitis A virus |
Like enteroviruses |
Like enteroviruses |
|
Formerly serotype 72 of the genus Enterovirus, now placed with the genus Hepatovirus |
Laboratory adapted hepatitis A virus strains can be grown in culture, but the wild type virus is more fastidious. |
By PCR, it has been shown in stools [Yotsuyanagi et al., 1996], sewage effluents, polluted waters [Tsai et al., 1994] and bivalve shellfish |
Diseases progresses from fever, headache, nausea and malaise to vomiting, diarrhoea, abdominal pain and jaundice Hepatitis A is self-limiting and rarely causes death, although patients may be incapacitated for several months. Complete recovery with long-term immunity from reinfection. Endemicin developing countries with most children seropositive by 6 |
Bivalve shellfish frequently implicated in outbreaks. |
Bivalve molluscs have consistently been proven to be an effective vehicle for the transmission of viral diseases (Lees, 2000). The high risk results from two facts: shellfish are filter feeders and many of them are consumed whole and raw or only lightly cooked. The risk is increased because many species are cultivated in near-coastal waters, where contamination with human sewage - which may contain high levels of viral particles - may easily occur. The largest known outbreak of hepatitis A occurred in Shanghai, China, in 1988, where almost 300 000 cases were traced to the consumption of clams harvested from a sewage-polluted area (Halliday et al., 1991; Tang et al., 1991).
In addition, shellfish harvested in contaminated areas may carry a mixture of viruses and consumers may be simultaneously contaminated with more than one viral strain (Lees, 2000). This probably contributes to the very high attack rates registered (sometimes up to 100 percent) and explains why, in some outbreaks, patients may suffer hepatitis after an initial gastro-enteritis (Halliday et al., 1991; Richards, 1985).
The need to re-evaluate the methodology used for the control of viral contamination is illustrated by the fact that in many outbreaks the harvesting areas, treatment processes and products sold complied with the current regulatory requirements established by the different countries involved (Fleet et al., 2000; Lees, 2000).
The development of methods for detection of viruses that can cause food-borne diseases has been concentrated on molluscan shellfish, due to their proven association with outbreaks. Viruses do not grow in the shellfish tissue, but need to infect some particular cells. Prior to the development of molecular methods the detection of viruses was dependent on the production of viral extracts suitable for inoculation into cell lines. Not all viruses, however, are culturable. A major problem with those that are is that the high cytotoxicity of the crude viral extracts obtained from contaminated shellfish makes them unsuitable for direct inoculation (Lees, 2000). A second problem is that no single cell line supports the growth of all culturable viruses, although some cells are useful for growing a variety of enteroviruses from environmental samples (Morris and Waite, 1980).
The detection of non-culturable enteric viruses has occasionally relied upon experimental infection of animals (Li et al., 1992) but has primarily depended upon visualization of viral particles by electron microscopy. While electron microscopy is still one of the main techniques for examination of clinical samples, its limitations and high technical demands make it inappropriate as a routine method for the examination of shellfish or environmental samples (Lees, 2000). More recently immunoassays have been used for food-borne disease outbreaks (Coulepis et al., 1985; Fleissner et al., 1989; Herrmann et al., 1985,1995; Moe et al, 1991; Monroe et al., 1993; Vizzi et al., 1996). However, the relatively high number of viral particles required for detection does limit the use of the technique for environmental samples and shellfish (Lees, 2000).
The application of molecular techniques to the detection of viruses has been possible thanks to the cloning and sequencing of hepatitis A (Cohen et al., 1987), hepatitis E (Bradley, 1995), Norwalk (Jiang et al., 1993) and related NLV viruses. Detection by probe hybridization has been hampered by the relatively high number of particles (thousands) required for detection, but PCR using consensus primers followed by sequencing of the amplified fragments, also called amplicons, has found wide application in detection and strain characterization (Lees, 2000).
To apply PCR, one has to know the sequences of DNA that are specific to the target organism. Although, theoretically, a single copy of the target in the reaction could end up as 240 copies in less than 3 hours, this is usually not the case: if the volume for analysis is 10 µl, then the minimum detectable level is 100 targets/ml. In practice, this is not feasible because there are many food components that inhibit the PCR reaction, and because the targets are not uniformly distributed in the sample. It is therefore advisable to first enrich the sample and/or to isolate the target (see below). Once the amplification is completed, the PCR products can be characterized by:
i. electrophoresis alone;
ii. restriction fragment analysis;
iii. single strand conformation polymorphism analysis;
iv. hybridization to a probe complementary to an internal sequence of the intended target; or
v. sequencing the amplicon.
Method i) only gives information about the apparent size of the fragment, while methods ii) to iv) and, of course, method v) also give some information about the sequence of the amplicon. It is also possible to perform several amplifications simultaneously in the same reaction tube (multiplex PCR), which can save time in the detection of several targets in one step.
PCR-based clinical assays have been reported for hepatitis A (Apairemarchais et al., 1995; Jansen, Siegl and Lemon, 1990), and E (McCaustland et al., 1991) viruses, astroviruses (Jonassen et al., 1995; Saito et al., 1995), rotaviruses (Gouvea et al., 1990; Xu, Harbour and McCrae, 1990) and enteric adenovirus (Tiemessen and Nel, 1996).
In addition to obtaining suitable primers to amplify only the targeted sequences, some of the problems encountered in applying PCR to the detection and identification of viruses in seafood are common to the application of this technique to all food matrices. These are elimination of inhibitors, concentration of the target, and detection of nonviable forms of the targeted organism. Other problems are specific to the nature of the virus as a target. Unlike bacteria, viruses do not multiply in the infected shellfish tissues and as less than 100 µl of sample is analysed, the viral particles have to be concentrated to reach detectable levels.
Design of suitable consensus primers has been more problematic for viruses whose strains show a high degree of diversity, such as NLV strains. The use of combinations of primer pairs to cover the spectrum of sequenced types has solved this problem and most of the known clinical NLV types can now be detected. For viruses that show little variation, such as hepatitis A, the application of PCR-detection has been straightforward (Lees, 2000).
Crude shellfish extracts seem to contain PCR inhibitors. The problem of elimination of inhibitors can be diminished, and simultaneously the target concentration increased, by extracting only the digestive organ of the shellfish, which is where viruses accumulate. This is followed by the usual purification and nucleic acid concentration steps included in most sample preparation protocols for PCR amplification (Lees, 2000). As in the case of bacteria, most of these studies have been made on artificially contaminated samples but it appears that the detection of viruses in naturally contaminated samples is more difficult. One reason may be that naturally contaminated samples have lower levels of the target sequence. Increasing the sensitivity of the amplification by nested PCR has helped to overcome this problem (Dore, Henshilwood and Lees, 1998; Green et al., 1998).
Target-recognition techniques can produce false positives due to cross-reactions with similar sequences of, usually, related organisms. In addition, as long as the targeted fragment of nucleic acid is not destroyed, non-viable forms of viruses will also be detected. The solution to the cross-reactions may be the design of more specific primers, increasing the stringency of the reaction, or sequencing of the obtained amplicon. Avoidance of the detection of non-viable forms is more complicated. The genome of most of these viruses is made up of RNA, which is more readily degraded than DNA, and is therefore quickly degraded by the digestive enzymes of the shellfish or the associated bacterial flora. Yet, some samples, artificially seeded with feline calicivirus (used as a model for Norwalk group viruses) that had been heat-treated to inactivate the viruses and subsequently failed to yield virus on culture, gave positive results by reverse transcriptase (RT)-PCR (Slomka and Appleton, 1998). Similar results have been obtained in the PCR-based detection of L. monocytogenes (see below) when the level of targeted sequence was high and the samples analysed immediately after the heat-destruction of the organism. The detection of non-viable forms is thus a problem that needs to be addressed and some researchers have proposed starting with an initial round of culture in cells followed by PCR-detection or a combination of immunological and molecular techniques. The first approach is not applicable to non-culturable viruses and will significantly lengthen the analysis time. Moreover, the analysis will be restricted to laboratories with facilities for culturing viruses in cell lines. The second approach, a combination of immunological and molecular techniques, may simultaneously enrich the sample, eliminate contaminants and collect only viable particles by immunological techniques, thus simplifying the sample preparation procedure for PCR detection. However, the quality of the analysis will be highly dependent on the quality and reactivity of the antibodies used.
One of the problems that molecular techniques based on PCR were not able to solve until recently was the quantification of the targeted sequence. Quantification of a target by PCR can now be achieved by competitive PCR and by real-time quantitative PCR (Holland et al., 1991).
Competitive PCR is time-consuming and troublesome, it requires the use of serial dilutions of the target sequence and a competitive sequence. Both must have similar efficiencies for amplification, but the assay is linear only over a short range of concentrations (Grove, 1999).
Real-time quantitative PCR is based on the accurate measurement of a fluorescent signal produced during amplification, the intensity of the fluorescence being proportional to the number of copies produced. The two pioneer companies manufacturing instruments for real-time quantitative PCR (Roche manufacture the Light Cycler, and Perkin Elmer/Applied Biosystems the 5700 and 7700 sequence detectors) use different technical principles, but in both cases the acquisition of data is almost instantaneous, as it takes place when the amplification is still in the exponential phase. The method is nicely described by Grove (1999): basically, the fluorescence is monitored until, after a particular cycle, it reaches a level above the background. That particular cycle is called the threshold cycle (Ct) and it is inversely proportional to the number of target sequences that were originally in the template: the higher the number of target copies, the less the number of cycles required for its detection and the shorter the detection time. The use of more than one fluorescent dye enables the performance of quantitative multiplex PCR: the detection and quantification of several targets in the same reaction. This technique has been used to detect hepatitis A virus (HAV) in shellfish at a level of 104 to 107 copies of HAV/gram (Arnal et al., 1999), (Norwalk-like virus) and hepatitis A virus in clams (Furuta et al., 2003).
For convenience, Huss (FAO, 1994) divided the pathogenic bacteria associated with seafood-borne diseases into two groups. The first contains bacteria naturally found in the aquatic environment, such as Clostridium botulinum, Vibrio spp., Aeromonas hydrophila, Plesiomonas shigelloides and Listeria monocytogenes. The more psychrotrophic L. monocytogenes and C. botulinum are more frequently found in Arctic and colder climates, while the more mesophilic Vibrio spp. are more frequently found in warmer environments. The second group consists of mesophilic bacteria that contaminate the aquatic environment from human or animal reservoirs such as Salmonella spp., Escherichia coli, Shigella spp. or Staphylococcus aureus. The degree of processing of seafood has implications for the associated risks: botulism, listeriosis and cholera are more frequently associated with consumption of smoked, fermented, salted and pickled products, which are usually ingested without further processing (FAO, 1994; Jay, 2000).
The United States General Accounting Office (1996a) targeted four bacteria as of major concern in food-borne diseases in the United States: E. coli O157:H7, Salmonella enteriditis, Listeria monocytogenes and Campylobacter jejeuni. It is interesting to note that three of the four - E. coli O157:H7, L. monocytogenes and C. jejeuni - were not recognized as food-borne disease causing agents only 20 years ago.
Gilbert, James and Sintchenko (1999) reviewed the use and limitations of molecular methods for the diagnosis of infectious diseases. There is no universal method that permits identification and typing of all bacteria, as each species requires specific conditions for growth as well as antibodies, primers or probes for detection. Due to its impact on fish products, L. monocytogenes has been chosen as an example in order to illustrate the methods of detection, identification and typing of bacteria.
There are two basic classical approaches to detection of the presence of any given micro-organism in a food product: direct plating from the food onto a selective agar medium and incubation of the product in a resuscitation and/or enrichment medium, followed by plating onto a selective agar medium.
The bacteria present in food matrices have usually been subjected to stressful conditions (cold storage, drying, freezing, smoking, heat shock, antibacterial substances, etc.) that may have killed many of them and injured another fraction. A few may have survived the treatment but require optimal conditions for a period of time in order to start growing again. Therefore, it is convenient to incubate them in a resuscitation medium first, followed by a medium selective for the targeted micro-organism.
Patel (1994) gives an account of the methods used in modern microbiology, all of which have been applied to the detection of a variety of micro-organisms, including L. monocytogenes. The introduction of membrane filtration techniques permitted the retention of bacteria in filters than can be placed directly on agar plates or soaked in nutrients which allow the growth of the micro-organisms on the filter. This procedure allows the filtration of large volumes of sample while eliminating inhibitors in the process. The filter can be transferred from one media to another without interfering with the growth, and it is possible to use electronic counters for automated counting.
Bacterial growth can also be measured by using automated electrical techniques. Metabolism and growth result in an increase in the number - and a decrease in the size - of charge-carrying molecules in the growth medium. These molecules are better electrical carriers. Electrical techniques estimate bacterial growth by measuring changes in the conductivity of the medium. They have been applied to routine analysis following the development of suitably selective media for relevant bacterial species and of computer technology to record, store and present the data to the operator.
Bacteria can be selectively removed from a medium by using immunomagnetic beads which are magnetic particles coated with streptavidin and antibodies specific for the target. Since the particles are magnetic, they will stick to the walls of the tube when in contact with a magnetic particle concentrator. The medium and non-targeted organisms can be pipetted away while the immunomagnetic particles, with the targeted pathogen, can be resuspended in a new medium or used directly for subsequent analyses such as PCR amplification.
Immunological techniques have also been used to develop test kits based on the same principle as the Jellet Rapid Test kit for the detection of biotoxins. The strips look very similar (with only differences in the shape and colour of the plastic cassette); they also claim to take about 20 minutes and commercial kits are already available to detect E. coli O157 and Campylobacter spp.
Molecular techniques are mainly based on the use of PCR. New genes are being characterized and sequenced every day, so the number of sequences available for PCR detection of micro-organisms is continuously increasing. Bacteria successfully detected by PCR include L. monocytogenes (targeting listeriolysin and iap genes); Campylobacter coli, C. jejuni (targeting flagellar protein genes) and other Campylobacter spp.; Clostridium botulinum (targeting the gene for the neurotoxin). E. coli, Salmonella typhimurium and Shigella flexineri were detected by specific PCR amplification of the lam B gene (which encodes the bacteriophage lambda receptor) and enterotoxigenic strains of E. coli by targeting the gene for the enterotoxin. As previously discussed, it is advisable to first enrich the sample and/or to isolate the target, such as by membrane filters or immunomagnetic beads - which concentrate it and at the same time eliminate the presence of background flora and food inhibitors - prior to amplification.
Real time quantitative PCR has been applied to some human pathogens in food matrices: about 2-6 cfu of S. typhimurium per PCR reaction were detected in chicken carcass rinses, ground beef, ground pork and raw milk by Chen et al., (1997 a, b); enterotoxigenic Clostridium perfringens in beef, pork, and chicken meat (Miwa et al., 1998). Targeting Shiga-like toxin genes, Witham et al., (1996) identified about 0.5 cfu of Shiga-like toxin I-producing E. coli per gram in ground beef after a 12 h enrichment in modified E. coli broth. A review of the application of this technology to dairy-borne pathogens has been published by Batt (1997).
Listeria monocytogenes is an ubiquitous Gram-positive bacteria. There are seven species of Listeria: L. monocytogenes, L. innocua, L. seeligeri, L. welshimeri, L. ivanovii, L. grayi and L. murrayi. Only L. monocytogenes is considered to be a pathogen of concern for humans (Ryser and Marth, 1991).
L. monocytogenes is found naturally in soils, sewage and freshwater sediments and is frequently carried in the gastrointestinal tract of animals and humans (Farber and Peterkin, 1991). The existence of asymptomatic carriers is well known and it has been estimated that between 2 and 6 percent of the human population are positive at any given time (Rocourt, 1994). The importance of L. monocytogenes as a food-borne human pathogen has only been recognized since the 1980s (Farber and Peterkin, 1991; Gellin et al., 1991). Between 85 and 95 percent of cases of listeriosis are food-borne (Buzby et al, 1996). Listeriosis can occur in healthy individuals, but the primary groups at risk are the immunocompromized, including the foetuses of pregnant women, the elderly, AIDS patients and those under medication to reduce their immune responses, such as cancer patients and organ transplant recipients. Listeriosis has a long incubation period, from one to several weeks (Ryser and Marth, 1991), making it very difficult to associate foodstuffs to sporadic cases of the diseases. The symptoms of the infection may vary from asymptomatic infection, to skin infections and flu-like symptoms, to miscarriage, stillbirth, sepsis, meningitis and death (Farber and Peterkin, 1991). The death rate in the United States has been estimated to be between 23 percent (Tappero et al., 1995) and 28 percent (Schwartz et al., 1988), except for a Californian outbreak caused by Mexican-style cheese, where it was almost 34 percent (Jay, 2000). Between 1986 and 1988, the mortality rate in the United Kingdom was between 51 percent and 44 percent for perinatal and adult cases respectively (McLauchlin, 1987).
Although listeriosis has not often been associated with seafood consumption (Ryser and Marth, 1991), shellfish and raw fish are thought to have played a role in an outbreak in Auckland, New Zealand in 1980 (Lennon et al., 1984). The outbreak lasted for 11 months, with 22 perinatal listeriosis cases registered, including 5 foetal and 1 liveborn deaths. Most of the isolates were serovar 1b and no cultures from foods were obtained.
L. monocytogenes has been isolated from retail frozen seafood in 9 of 12 countries in a study by Weagant et al. (1988) and from seafood in Taiwan (Wong, Chao and Lee, 1990). Again, although the long incubation period of the disease makes it difficult to trace sporadic cases back to remaining food items, Loncarevic et al. (1998) found that L. monocytogenes isolated from two clinical cases and from two fish products (vacuumpacked smoked rainbow trout and gravad salmon) in Sweden were indistinguishable. Therefore, fish products cannot be disregarded as a source of illness. The techniques used by these authors were phage typing and restriction enzyme analysis (REA) with pulse-field gel electrophoresis (PFGE) of the restricted fragments. Similarly, a high prevalence of L. monocytogenes serogroup 1/2 in cold smoked and gravad rainbow trout and salmon has been reported in Norway by Rørvik, Caugant and Yndestad (1995). These authors found that the most common electrophoretic type among L. monocytogenes serogroup 1/2 strains, obtained from vacuum-packed smoked salmon and from a processing plant, was ET6. Strain ET6 has also been isolated from sporadic cases of human listeriosis in an outbreak in Norway in 1995 (Rørvik, Caugant and Yndestad, 1995). Curiously, and in spite of its ubiquitous nature, L. monocytogenes does not seem to be detectable in tropical seafood products (Ben Embarek, 1994; Fuchs and Surendran, 1989; Muntiningish and Sunarya, 1998).
L. monocytogenes can contaminate estuarine environments though sewage, processing and agricultural effluents. The relatively low salt content in this environment does not affect its survival capacity. Up to 11 percent of unprocessed shrimps in the Gulf of Mexico were L. monocytogenes positive, as well as about 1.5 percent of retailed fresh water fish. The Food and Drug Administration found that 26 percent of frozen seafood products from various countries that were found to be positive were shrimp (raw and cooked), cooked crab meat and surimi.
Between 9 percent and 28 percent of the samples of ready-to-eat shrimps, crab and smoked fish analysed in several surveys contained L. monocytogenes (Dillon, Patel and Ratman, 1994; Farber and Peterkin, 1991; Rocourt and Bille, 1997). Harvey and Gilmour (1993) found that 18.3 percent of 513 food samples examined in a survey in Northern Ireland contained L. monocytogenes. While 229 of the 513 food items had received cooking presumed sufficient to eliminate Listeria, 12.2 percent of them were L. monocytogenes positive. In Norway, Rørvik and Yndestad (1991) examined 382 samples of retail food items (imported soft cheese, raw chicken, minced meat, fermented sausages, vacuum-packed processed meat products, smoked salmon, peeled shrimps, raw minced fish) and 78 carcass samples (sheep, pig, cattle) and found 16.3 percent of them to be positive. L. monocytogenes was most frequently isolated from raw chicken, sporadically from soft cheese, shrimps, processed meat products and smoked salmon, yet not from carcasses and fermented sausages. In southern Finland, 20 percent of 110 ready-to-eat vacuum packed fish products (hot- and cold-smoked and salted fish) were found to be L. monocytogenes positive in a survey carried out by Johansson et al. (1999). In a survey of Japanese retail foods, Inoue et al., [2000] found between 12.2 and 37 percent of minced meats, 5.4 percent of smoked salmon samples and 3.3 percent of ready-to-eat seafood positive for L. monocytogenes, although they did not find the bacterium in any of 285 vegetable samples examined.
Often different strains are isolated from raw materials and final products. Some studies show that L. monocytogenes can colonize the processing environment (utensils, brines, etc.) (Destro, Leitao and Farber, 1996; Giovannacci et al., 1999; Lawrence and Gilmour, 1995; Martinez et al., 2003a; Rørvik, Caugant and Yndestad, 1995, Rørvik et al., 1997). Colonization of the processing environment (FAO, 1999; Giovannacci et al., 1999; Johansson et al, 1999; Rørvik et al., 1995, 1997) as well as job rotation among departments (Rørvik et al., 1997) have been established as primary mechanisms for contamination of some products.
Detection of L. monocytogenes
Traditional detection methods for L. monocytogenes involved a cold enrichment procedure followed by plating (see Ben Embarek, 1994 for a review; Biester and Scharte, 1939). These methods may require months for detection of the bacterium. Newer methods use enriched selective media, shorter time (about 48 h) and higher temperature (30-37o C) (see Loncarevic, 1998 and references therein). The selection of the most suitable medium for recovery of L. monocytogenes from food matrices depends on the food type, the strain infecting the product, the level of infection and the degree of injury to the bacterium (Loncarevic, 1998). The lack of an optimal procedure is reflected by the fact that different official bodies have chosen different protocols (see Loncarevic, 1998; Lovett, 1988; McClain and Lee, 1988; NMKL, 1990). It has also been shown that the enrichment and plating media may favour or suppress the growth of different strains of L. monocytogenes to different degrees (Loncarevic, Tham and Danielsson-Tham, 1996, 1997).
All virulent strains of L. monocytogenes produce a specific substance responsible for the b-haemolysis of erythrocytes and the destruction of phagocytic cells that engulf them. This substance has been called listeriolysin O (LLO) and is homologous with streptolysin O (SLO) and pneumolysin (PLO) (Mengaud et al., 1987, 1988). Colonies of suspected Listeria spp are identified by their ability to produce b-haemolysis on blood agar and by biochemical tests (Rocourt, Schrettenbrunner and Seeliger, 1983). The biochemical identification has been simplified by the introduction of a rapid test strip identification system that is claimed to reduce the time needed for conventional identification of suspected colonies to about 24 h (Bille et al., 1992).
The most recent methods are based on the PCR-based detection of DNA sequences specific for L. monocytogenes. Virulence or invasion genes, such as the listeriolysin O (hly) gene were used by Agersborg, Dahl and Martinez (1997), Furrer et al. (1991), Golsteyn Thomas et al.(1991) and Niederhauser et al. (1992), while the invasion-associated protein (iap) gene was selected by Agersborg, Dahl and Martinez (1997) and Jaton, Sahli and Billie (1992).
Since only a few µl are used for the amplification (typically 10-50 µl), and absence of inhibitors is required, it is necessary to design protocols both to increase the concentration of the target organism and to eliminate those inhibitors. Past experience has shown that it is necessary to incubate the food samples for at least 48h to be able to detect low levels of L. monocytogenes (1 to 5 bacteria per 200 ml broth) and simultaneously avoid false positives due to the presence of high levels of non-viable
L. monocytogenes. Incubations of only 24h can render false negative and false positive results respectively (Agersborg, Dahl and Martinez, 1997). Unfortunately, the use of immunomagnetic beads, which could significantly reduce the time of sample preparation, has not shown promising results (Skjerve, Rørvik and Olsvik, 1990; Uyttendaele, van Hoorde and Debevere, 2000).
After PCR amplification, hybridization of the amplicon with a probe recognizing an internal sequence, has been shown by Xiaoming et al. (2000) to be effective, not only to confirm that the correct target had been amplified, but also to increase the sensitivity of detection. These authors successfully used this approach for the simultaneous detection of Salmonella typhimurium and L. monocytogenes from pure bacterial mixtures and seeded milk.
Real-time quantitative PCR has also been applied to the detection and quantification of L. monocytogenes. We have not found published works using real-time PCR to detect L. monocytogenes in seafood, but it has been successfully applied to other food and environmental samples. Thus, Nogva et al., (2000) could detect approximately 6 to 60 CFU per amplification targeting the listeriolysin O gene (hly A) in pure cultures, water, skim milk, and unpasteurized whole milk and using paramagnetic beads for the isolation of bacteria from the media. The iap gene was the target selected by Hein et al., (2001) who had a limit of detection of 6 copies of the gene per reaction. Comparison of detection levels in cheeses showed that 0.04-0.4 and 4 CFU per gram could be detected respectively in homogenates of mozzarella and crescenza but one needed 40 CFU per gram to detect the pathogen in ricotta cheese (Longhi et al., 2003). Real-time PCR has also been used to detect three pathogens simultaneously: fresh vegetables (green cabbage, broccoli, cauliflower and cilantro) were washed with artificially contaminated water containing E. coli O157:H7, S. typhimurium and L. monocytogenes: the real-time PCR analysis was able to confirm the predicted levels of 1 to 10 cells/ml for E. coli O157:H7 and S. typhimurium and at 1000 cells/ml for L. monocytogenes (Bhagwat, 2003).
TABLE 3
Serovars of Listeria spp. According to [Seeliger and Jones, 1986]. ()a, not always present.
|
Listeria spp |
Serovars |
O antigens |
H antigens |
||||||||||||||
|
monocytogenes |
1/2a |
I |
II |
(III)a |
|
|
|
|
|
|
|
|
|
A |
B |
|
|
|
monocytogenes, seeligeri |
1/2b |
I |
II |
(III) |
|
|
|
|
|
|
|
|
|
A |
B |
C |
|
|
monocytogenes |
1/2c |
I |
II |
(III) |
|
|
|
|
|
|
|
|
|
|
B |
|
D |
|
monocytogenes |
3a |
|
II |
(III) |
IV |
|
|
|
|
|
|
|
|
A |
B |
|
|
|
monocytogenes |
3b |
|
|
(III) |
IV |
|
|
|
|
|
(XII |
XIII) |
|
A |
B |
C |
|
|
monocytogenes |
3c |
|
|
(III) |
IV |
|
|
|
|
|
(XII |
XIII) |
|
|
B |
|
D |
|
monocytogenes |
4a |
|
|
(III) |
|
(V) |
|
VII |
|
IX |
|
|
|
A |
B |
C |
|
|
monocytogenes, innocua |
4ab |
|
|
(III) |
|
V |
VI |
VII |
|
IX |
X |
|
|
A |
B |
C |
|
|
monocytogenes |
4b |
|
|
(III) |
|
V |
VI |
|
|
|
|
|
|
A |
B |
C |
|
|
monocytogenes, seeligeri |
4c |
|
|
(III) |
|
V |
|
VII |
|
|
|
|
|
A |
B |
C |
|
|
monocytogenes, seeligeri |
4d |
|
|
(III) |
|
(V) |
VI |
|
VIII |
|
|
|
|
A |
B |
C |
|
|
monocytogenes |
4e |
|
|
(III) |
|
V |
VI |
|
(VIII) |
(IX) |
|
|
|
A |
B |
C |
|
|
ivanovii |
5 |
|
|
(III) |
|
(V) |
VI |
|
(VIII) |
|
X |
|
|
A |
B |
C |
|
|
monocytogenes |
7? |
|
|
(III) |
|
|
|
|
|
|
XII |
XIII |
|
A |
B |
C |
|
|
innocua, welshimeri |
6a (4f) |
|
|
(III) |
|
V |
(VI) |
(VII) |
|
(IX) |
|
|
XV |
A |
B |
C |
|
|
innocua, welshimeri, seeligeri |
6b (4g) |
|
|
(III) |
|
(V) |
(VI) |
(VII) |
|
IX |
X |
XI |
|
A |
B |
C |
|
|
grayi |
|
|
|
(III) |
|
|
|
|
|
XII |
|
XIV |
|
E |
|
|
|
|
murrayi |
|
|
|
(III) |
|
|
|
|
|
XII |
|
XIV |
|
E |
|
|
|
Typing of L. monocytogenes
Strain identification is critical to determining the source or sources of infection - the index case, the product originating the outbreak, or the focus of contamination in a factory - as well as the routes of spreading a given infection.
Serotyping is based on the ability of the bacteria to agglutinate antibodies anti-somatic and anti-flagellar antigens. The seven species of Listeria possess antigens that give rise to 17 serovars: the somatic O antigens give 15 serovars and there are 5 flagellar antigens H (Table 3). One of the problems with serotyping is that some of the 13 serovars of the pathogenic L. monocytogenes are shared by L. innocua and L. seeligeri. The other is that most isolates from food and clinical cases belong to serotypes 1 and 4, with serogroup 1/2 more frequently encountered in food isolates and serogroup 4 in clinical isolates (Farber and Peterkin, 1991). Thus, the low discrimination of serotyping makes it of limited value for epidemiological studies.
Multilocus enzyme electrophoresis (MEE) estimates the genomic relationship among isolates by determining the relative electrophoretic mobilities of a set of water-soluble polymorphic bacterial enzymes, after starch electrophoresis and specific enzymatic staining techniques (Selander et al., 1986). The result of the application of this technique to each bacterial isolate and enzyme is called an electromorph. The combination of electromorphs for all the enzymes is called the electrophoretic type (ET). Typically, MEE typing requires the analysis of between 15 and 25 polymorphic enzymes (Jay, 2000). Differences in the electrophoretic mobility of allelic variants are dictated by differences in the amino acid sequences and therefore in the nucleotide sequence. Since some enzymes may be polymorphic among different strains of the same species, MEE can be used in epidemiological studies, giving additional information to that obtained by serotyping alone. All isolates can be typed by MEE and there is good correlation between MEE types and serotypes (Jay, 2000).
Phage-typing classifies bacterial isolates according to the phages that can lyse them. Phages are viruses that can infect bacteria, and were first described in 1945 (Jay, 2000). They contain double stranded DNA and belong to two groups: Siphoviridae (phages with non-contractile tails) and Myoviridae (phages with contractile tails). Phage-typing is usually considered to show good reproducibility (Audurier et al., 1984; Loessner and Busse, 1990; McLauchlin, Audurier and Taylor, 1986) but no correlation seems to exist between lytic patterns and species and serovar types (see Loessner and Busse, 1990). One major disadvantage of this technique is that at present there are not enough phages to lyse all L. monocytogenes isolates (Audurier and Martin, 1989).
For typing by Restriction Enzyme Analysis (REA), the whole isolated bacterial chromosome is digested by restriction endonucleases. The resulting high molecular mass fragments (from a few kilo-bases to over 10 mega-bases) are resolved by agarose-gel electrophoresis or pulsed-field gel electrophoresis (PFGE), depending on their size (Southern and Elder, 1995). Since the restriction enzymes cut the double stranded bacterial DNA at specific sequences, the number and size of the resulting fragments will depend on the nucleotide sequence. The analysis will therefore reflect genomic differences among isolates. REA alone has been used to characterize strains of L. monocytogenes isolated from listeriosis outbreaks (Baloga and Harlander, 1991; Nocera et al., 1990) and shown that isolates from major human epidemics exhibit unique restriction patterns (Wesley and Ashton, 1991). The combination of low-frequency cutting enzymes for REA with PFGE has made the patterns easier to interpret and has proved to be a highly discriminatory technique (Brosch, Buchrieser and Rocourt, 1991; Brosch, Chen and Luchansky, 1994; Carrier et al., 1991).
Ribotyping is based on the characterization of isolates by identifying polymorphisms in the ribosomal RNA genes. The genes coding for the 16S and 23S rRNA contain some hypervariable regions suitable for genus and species identification, but there are also some highly conserved regions that permit the use of rRNA from E. coli as a universal probe. Moreover, since most bacteria contain multiple ribosomal operons - an operon is a genetic regulatory system found in bacteria and their viruses in which genecoding for functionally related proteins are clustered along the DNA - ribotyping provides patterns complex enough to be suitable for subtyping.
Ribotyping has been applied to epidemiological investigations of L. monocytogenes and other pathogens, both in sporadic cases and in outbreaks (Baloga and Harlander, 1991; Graves et al., 1991, 1994; Grimont and Grimont, 1986; Nørrung and Gerner-Smidt, 1993; Stull, LiPuma and Edlind, 1988). It is a reproducible typing method but it usually fails to discriminate between 4b serovars (Baloga and Harlander, 1991; Nocera et al., 1993; Saunders, Ridley and Taylor, 1989; Wesley et al., 1990). These are most frequently associated with sporadic and epidemic listeriosis (Pinner et al., 1992; Schuchat et al., 1991; Schuchat, Swaminathan and Broome, 1991).
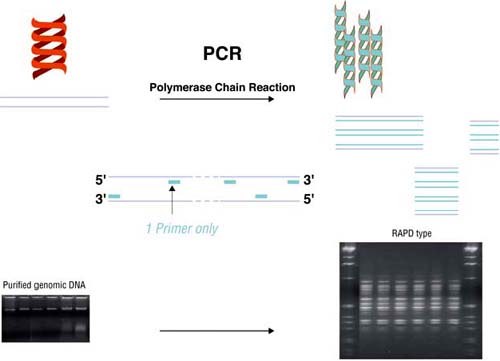
Random Amplification of Polymorphic DNA (RAPD) (Welsh and McClelland, 1990; Williams et al., 1990), is a PCR amplification, under non-stringent conditions, with arbitrary - usually short - primers (Figure 2). RAPD analysis has been used to type isolates of L. monocytogenes (Lawrence and Gilmour, 1995; MacGowan et al., 1993; Mazurier et al., 1992; Mazurier and Wernars, 1992). By using the appropriate primers, RAPD is a very discriminating technique because it targets many different loci simultaneously. Moreover, its discriminatory power can easily be increased by using different primers, since the supply of arbitrary primers is practically unlimited (Figure 3), and all genomes can be typed. However, the results of RAPD do not always agree with other typing methods. Strains of different serotypes may occasionally share the same RAPD-type (Destro, Leitao and Farber, 1996; Martinez et al., 2003a; Mazurier and Wernars, 1992), and the results of RAPD and phagetyping have not always shown a correlation (Mazurier et al., 1992).
|
FIGURE 2
Random Amplification of Polymorphic DNA (RAPD) is based on the non-stringent amplification of genomes by using arbitrary, short primers. The number and length of the amplicons produced depend on the loci that are complementary to the sequence of the primer and therefore dependant on the primer-DNA combination. The amplicons produced are separated and visualized, usually by agarose gel electrophoresis, staining and ultraviolet light, and a more or less complex pattern - the RAPD-type - is produced. |
|
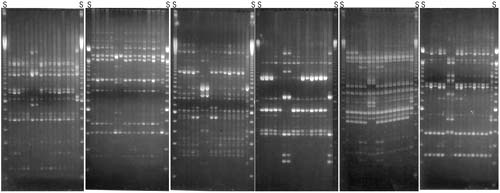
FIGURE 3
Each gel is the result of typing with a different primer. Each lane, in each gel, contains a different isolate. All gels contain the same isolates in the same order. S, lanes containing 100 bp ladder molecular mass standards. The 5th gel shows distortions that took place during the electrophoretic run. |
In a recent study, RAPD analysis has been used to examine the genetic relationship among 432 strains of L. monocytogenes isolated from clinical and veterinary cases of listeriosis, dairy, vegetable, meat- and fish-based food items, environmental samples and samples collected from one transport terminal, one poultry processing company and four Atlantic salmon processing plants (Martinez et al., 2003a). Great genetic variability among the isolates of L. monocytogenes was found, and none of the isolates from food products had the same RAPD fingerprint as isolates from human patients; so no particular food commodity could be linked to clinical cases in this work. Further, there was also great genetic variability within strains isolated from food processing environments (even within the same company) and in one plant the presence of some strains had persisted for years. The work did not support the presence of strains (as defined by RAPD typing) better suited for survival in specific food types or food processing environments, indicating that although differences may be found in the virulence of individual strains, all L. monocytogenes must be treated as potentially harmful.
The World Health Organization (WHO) sponsored an international collaborative study to evaluate methods for subtyping of L. monocytogenes (Bille and Rocourt, 1996). The methods tested, number of laboratories engaged, isolates and the results and conclusions of the trial according to the authors are shown in Table 4. No single method was found to be optimal to type all isolates used in the exercise.
One issue of major concern is the increase in the number of bacterial strains that are developing resistance to antibiotics, especially to several antibiotics simultaneously (multi-resistance). Resistance to antimicrobials existed before the introduction of antibiotics for the treatment of human and animal infections. The increase in the number of resistant strains, as well as in the number of antibiotics to which they show resistance, is attributed to their being selected and their dissemination enhanced, primarily due to the misuse and abuse of these drugs for treatment and prophylactic purposes. There is a genetic basis for resistance to antimicrobial substances. Susceptible strains may acquire the genes coding for the resistance mechanisms by conjugation (between bacteria of the same or similar species), transformation (uptake of DNA from unrelated species), or transduction (the transfer of DNA via a bacteriophage) (European Commission, 2000 and references therein; McDonnel and Russell, 1999).
Antibiotic abuse may unfortunately not be the only problem: the use of cleaning and disinfectant agents in hospitals, by the food industry, and at private households, seems to have had the undesirable side-effect of permitting survival and therefore selection of strains resistant to disinfecting agents.
Disinfectants based on quaternary ammonium compounds (QAC, such as benzalkonium chloride (BC), cetylpyridinium chloride, cetrimide, etc.) are commonly used in the food industry because they are non-corrosive and have low toxicity. Regrettably, there seem to be similarities between bacterial resistance to antibiotics and to biocides: the spread and maintenance of QAC-resistant genes in staphylococci isolated from clinical environments and the food industry may be due to the selective pressure caused by the use of cationic biocides (Stickler and King, 1992). Gram-negative bacteria that have developed resistance to cationic biocides may also be resistant to some antibiotics (Russell, 1997, 2000). Outer membrane changes are believed to be responsible for the non-specific increase in resistance; but also increased tolerance to disinfectants can be due to the presence of energy-dependent efflux pumps in the cell membrane (Gillespie, May and Skurray, 1986; Noguchi et al., 1999) and associated with the qacA/B gene system in staphylococci, (Russell, 2000). Among staphylococci, the genes encoding multidrug exporter proteins can be divided into two families based on DNA homologies and phenotypic properties (Littlejohn et al., 1992; Rouch et al., 1990): the qacA-qacB (QACs resistance) and smr (multidrug resistance) families, the former having a wider resistance phenotype (Leelaporn et al., 1994). Known resistance genes to QACs described in clinical staphylococci are generally plasmid borne and widely distributed in the environment (Lyon and Skurray, 1987; Mayer et al., 2001), and qac resistance determinants have been found together with antibiotic resistance genes encoding resistance to gentamicin (GEN), trimethoprim (TMP), penicillin, kanamycin (KAN), and to bramycin on some plasmids (pST6, pSK4, and pSK41) and transposons (Tn 552 and Tn 4002) (Berg et al., 1998; Leelaporn et al., 1995; Lyon and Skurray, 1987; Sidhu et al., 2001).
TABLE 4
Results of the WHO-sponsored international collaborative study to evaluate methods for subtyping of L. monocytogenes (Bille and Rocourt, 1996). Each participant laboratory received a set of 80 L. monocytogenes strains which contained 22 groups of epidemiologically linked isolates and 11 pairs of duplicate strains.
|
Method and reference |
Results and conclusions by the authors |
|
Serotyping |
6 participant laboratories. Complete agreement between the six laboratories on 49 (61.3%) strains (21 serovar 1/2a and 28 serovar 4b strains) The intralaboratory reproducibility ranged from 82 to 100%, with a medium value of 91%. Reproducibility of serotyping L. monocytogenes strains according to serovar varied from 33.3 to 100% for serotypes 3b and 1/2a, respectively, with serovar 4b (x) being incorrectly identified in all six laboratories. Serotyping of L. monocytogenes is easy and simple and is a useful prerequisite for other finer and more discriminatory typing methods. Problems may however, be encountered mainly with the flagellar antigenic factors. There is a need, therefore, for preparing antisera of good quality from which efficient antigenic factors can be obtained. |
|
Pulsed-field gel electrophoresis (PFGE) Brosch et al., [1996] |
4 participant laboratories. The endonucleases ApaI and SmaI yielded between 21 and 28 digestion profiles (REDP). AscI, used in addition only in laboratory A, displayed 21 REDP. The combination of ApaI, SmaI or AscI REDP established 25 to 33 genomic groups depending on the laboratory and the number of viable strains. Agreement of typing data among the four laboratories ranged from 79 to 90%. Most of the epidemiologically related strains were correctly identified by the four groups of investigators. i.e., most related strains were placed into the same genomic groups by all four laboratories. In general this study reconfirmed that PFGE is a very accurate and reproducible method for fine structure comparison and molecular typing of L. monocytogenes. |
|
Random amplification of polymorphic DNA (RAPD) Wernars et al., [1996] |
6 participant laboratories. Using three different 10-mer primers the median reproducibility of the RAPD-results obtained by the six participants was 86.5% (range 0-100%). Failure in reproducibility was mainly due to results obtained with one particular primer. The number of epidemiological groups found to be homogeneous varied from 1-22 (median 16). However, for some groups an inhomogeneity was found by the majority of participants. The overall correlation between the results from the different participants ranged from 32 to 85%. |
|
High-frequency endonuclease (REA) typing Gerner-Smidt et al., [1996] |
2 participant laboratories. They used two restriction enzymes each. The enzymes were EcoRI, Hae1II, HhaI, and its isoschizomer Cfo1. EcoRI was the least discriminatory. The profiles generated by Hha1 and CfoI were not fully stable for some closely related isolates. The size and the number of restriction bands generated by Hha1 in one laboratory and Cfo1 in another laboratory were directly comparable, indicating that REA may be used as a definitive typing method for L. monocytogenes if the protocol is standardized. The stability of the REA-types needs further elucidation in order to establish firm differentiation criteria for comparison of isolates. |
|
Multilocus enzyme electrophoresis (MEE) Caugant et al., [1996] |
7 participant laboratories. Each laboratory used its own protocol. The number of enzymes that were assayed by the laboratories ranged from 8 to 23, and the total number of identified electrophoretic types (ETs) varied between 14 and 25. The discriminatory power of the method (Simpson’s index of diversity) ranged from 0.827 to 0.925. This relatively low discriminatory power is a consequence of a somewhat low genetic diversity of L. monocytogenes compared to other bacterial species. Efforts should be pursued to standardise the method in order to improve the intra- and inter-laboratory reproducibility. |
|
Phage-typing McLauchlin et al., [1996] |
6 participant laboratories. Phage typing was performed using an international phage set in five laboratories and phage sets unique to two laboratories. Testing of cultures sent in duplicate showed similar levels of reproducibility to those previously reported. Analysis of results from groups of epidemiologically related cultures showed a high level of agreement in all laboratories. Patterns of phage susceptibility were relatively stable on retesting strains in the same laboratory after long periods of time. However, there was limited comparability between results obtained from testing the same cultures using the same phages in different laboratories. It is recommended that the phages in the international set be reviewed, and that better inter-laboratory reproducibility may be achieved by standardization of phage suspensions, propagation strains and methodology, together with the use of centrally propagated phages. |
|
Restriction fragment length polymorphism (RFLP) analysis using ribotyping and Southern hybridization with two probes from L. monocytogenes DNA Swaminathan et al., [1996] |
7 participant laboratories. No standardized protocols. Six laboratories performed ribotyping with EcoRI to restrict the DNA and ribosomal RNA or DNA as the probe for Southern hybridizations. The seventh laboratory used Ncil to restrict the DNA, and two probes, one randomly cloned and the other containing repeat sequences cloned from L. monocytogenes DNA. The overall discriminating power of ribotyping (Simpson’s index of diversity) ranged from 0.83 to 0.88 for the six laboratories. The discriminating power of the combination of two probes used by lab 7 was 0.91. Ribotyping and the cloned probes used by lab 7 discriminated poorly between serotype 4b strains. Neither method identified three atypical strains (identified by other subtyping methods) included in three apparently epidemiologically related groups. Ribotyping did not discriminate between strains of serotypes 4b and 4b(X) in one epidemiologically related group of strains; one cloned probe used by lab 7 discriminated between these strains. Intra-laboratory reproducibilities for the seven laboratories ranged from 80.0 to 100%. Inter-laboratory reproducibilities were generally very good considering that no attempt was made to standardize protocols used by the participants. |
Recently, Sidhu et al. (2002) found that the frequency of resistance to several antibiotics was significantly higher among staphylococci resistant to the QAC-based disinfectant benzalkonium chloride (BC) than among strains sensitive to it. The same group of researchers (Anthonisen et al., 2002) showed that various staphylococcal species able to colonize human and animal hosts can exchange genetic material involved in resistance to antibiotics and disinfectants, and they warned that the lateral spread of resistance genes between different species may be facilitated by the generation of large multiresistance plasmids and the subsequent interspecies exchange.
Gene transfer both between different genera and between species within a genus may occur with the possible spread of resistance genes to pathogenic strains (see Im et al., 1996). Thus, clinical strains of QAC-resistant Staphylococcus aureus have been isolated in several countries from clinical environments (Leelaporn et al., 1994) and from food products and food processing environments (Heir, Sundheim and Holck, 1995, 1998). One of the plasmids that confers QAC-resistance, called smr (small multidrug resistance), has been found in enterococci and staphylococci (Sasatsu et al., 1995).
The commonly used household disinfectant pine oil has also been reported to induce the development of resistant strains. Moken, McMurray and Levy (1997) found that mutants of E. coli selected for resistance to pine oil overexpressed the marA gene and showed resistance to multiple antibiotics (tetracycline, ampicillin, chloramphenicol and nalidixic acid) and viceversa: antibiotic-selected Mar mutants that also overexpressed marA were resistant to pine oil. When pine oil was used to select resistant mutants of S. aureus it was found that the mutants had reduced susceptibility to the cell-wall active antibiotics vancomycin and oxacillin (Price et al., 2002).
Consequently, it seems that the acquisition of genes conferring resistance to disinfectants may also help the bacteria to tolerate some antimicrobial substances. The opposite, on the other hand, may not always be the case: as mentioned above, antibiotic-selected Mar mutants were resistant to pine oil (Moken, McMurray and Levy, 1997) but Kucken, Feucht and Kaulfers (2000) found that multiple antibiotic-resistant Gram-negative bacteria are not necessarily more resistant to quaternary ammonium compounds (benzalkonium chloride and cetyltrimethylammonium bromide). Similar results were obtained by Rutala et al. (1997) who reported that development of antibiotic resistance did not appear to be correlated with increased resistance to phenol and QAC.
Plasmid, and also some chromosomal genes are considered to be involved in the development of resistance to disinfectants and antibiotics: the expression of the E. coli chromosomal TehA genes confer to the bacterium resistance to antiseptics and disinfectants similar to that achieved by multidrug resistance efflux pumps (Turner, Taylor and Weiner, 1997).
The Opinion of the Scientific Steering Committee on Antimicrobial Resistance - 28 May 1999 (European Commission, 2000) indicates very clearly the need to reduce the overall use of antimicrobials. This implies elimination of unnecessary and improper use, more precise diagnoses of infectious agents, and monitoring of antimicrobial resistance. In order to design sensible disinfection procedures, Langsrud and Sundheim (1997) suggested the convenience of alternating the use of QACs with chlorine, phenolics, and alkylaminoacetate, to avoid the build-up of resistant strains. In addition there is a need for more rapid and accurate microbiological diagnostic tools to identify the resistance genes for the different antimicrobials carried by the strain under examination. The PCR (particularly multiplex PCR, to detect several resistances simultaneously) can be the technique of choice to detect those targets in a fast and specific manner once the different genes conferring resistance to antimicrobials and/or disinfectants are sequenced. Known sequences are given in some of the listed publications cited above, but new sequences are continuously being published and added to the available databases.
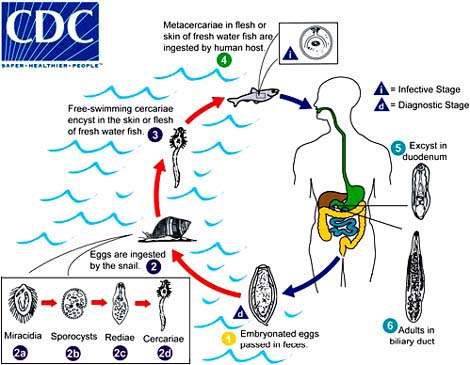
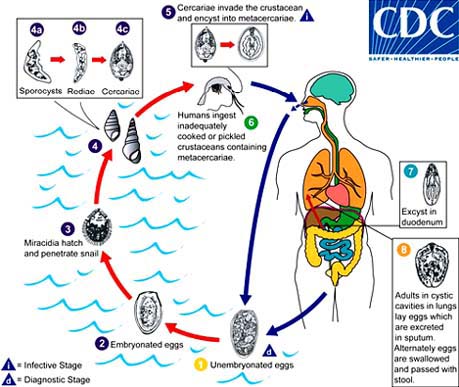
Protozoa, platyhelminths and nematodes can also cause food-borne diseases. Of special relevance to fish products are trematodes and some nematodes, whose taxonomical classification is shown in Table 5. Trematodes, or flukes, infect the liver, lungs or blood of mammals. Paragonimiasis is caused by species of the genus Paragonimus. P. westermani is predominant in Asia, but is also found in Africa and South and Central America, while P. kellicotti is more frequent in North and Central America. P. westermani is a lung fluke. Clonorchiasis, caused by Clonorchis (Opisthorchis) sinensis, the Chinese liver fluke, causes oriental biliary cirrhosis. Opisthorchiasis is caused by Opisthorchis felineus and O. viverrini. Clonorchis and Opisthorchis are very similar both morphologically and in their life cycles (Figure 4). The infections are mostly contracted from the ingestion of raw or improperly cooked crabs or fish.
Paragonimiasis is acquired by ingesting crustaceans infected with metacercariae which hatch and bore their way as young flukes through the walls of the duodenum and then move to the lungs, where they become enclosed in connective tissue cysts (Figure 5). Golden brown eggs may appear in sputum 2 to 3 months later. Clonorchiasis is due to the ingestion of fish containing the metacercariae: the cyst wall dissolves in the intestine, the young flukes emerge and migrate through the body to the bile ducts of the liver (Figure 4). C. sinensis causes, among other complaints, cirrhosis and liver cancer. Both paragonimiasis and clonorchiasis are diagnosed by demonstrating the presence of eggs in sputum, stools or duodenal fluid. There are some ELISA tests available (for the infection, not for the parasite) but they give cross-reactions with infections caused by related species.
TABLE 5
Taxonomical classification of some parasites causing food-borne diseases
|
Phylum |
Platyhelminhes |
||||||
|
|
Class |
Trematoda (flukes) |
|||||
|
|
|
Subclass |
Digenea |
||||
|
|
|
|
Order |
Plagiorchiata |
|||
|
|
|
|
|
Family |
Troglotrematidae |
||
|
|
|
|
|
|
Genus |
Paragonimus |
|
|
|
|
|
|
|
|
Species |
P. westermani in Asia, |
|
|
|
|
Order |
Opisthorchiata |
|||
|
|
|
|
|
Family |
Opisthorchiidae |
||
|
|
|
|
|
|
Genus |
Opisthorchis |
|
|
|
|
|
|
|
|
Species |
O. felineus in Kazakhstan, Russian Federation and Ukraine |
|
|
|
|
|
|
|
Species |
O. viverrini in Thailand and Lao People’s Democratic Republic |
|
|
|
|
|
|
Genus |
Clonorchis |
|
|
|
|
|
|
|
|
Species |
C. sinensis in China |
|
|
Class |
Cestoidea |
|||||
|
|
|
Subclass |
Eucestoda (tapeworms) |
||||
|
|
|
|
Order |
Pseudophyllidea |
|||
|
|
|
|
|
Family |
Diphyllobothriidae |
||
|
|
|
|
|
|
Genus |
Diphyllobothrium |
|
|
|
|
|
|
|
|
Species |
D. latum in USA, Scandinavia |
|
Phylum |
Nematoda |
||||||
|
|
Class |
Secernentea (Phasmidia) |
|||||
|
|
|
|
Order |
Ascaridida |
|||
|
|
|
|
|
Genus |
Ascaris |
||
|
|
|
|
|
Subfamily |
Anisakinae |
||
|
|
|
|
|
Genus |
Anisakis |
||
|
|
|
|
|
Genus |
Pseudoterranova (Phocanema) |
||
|
FIGURE 4
|
|
FIGURE 5
|
Anisakiasis is caused by the ingestion of fish flesh infected with nematodes: either Anisakis simplex (herring- or whaleworm) or Pseudoterranova (formerly Phocanema) decipiens (cod- or sealworm). Both parasites have several intermediate hosts and generally more than one definitive host. Humans are not the final host for either of them, and infections occur upon ingestion of fish containing molting larva from the second to the fourth stage. The nematodes do not mature in humans and symptoms are the result of activities of the juveniles. A. simplex is usually more harmful than P. decipiens because it can penetrate the mucosal lining while most P. decipiens are passed in faeces, coughed up or vomited after irritating the mucosa. The diagnosis of this condition is complicated by the absence of eggs from faeces. Larvae can be seen in the intestinal tract by endoscopy and the affected tissue can be removed surgically (Jay, 2000).
Food-borne parasitic diseases affect millions of people world-wide. About 700 million people are considered to be at risk of contracting food-borne trematode infections (FBT) and 40 to 50 million are believed to be infected by one or more trematode parasites. (FAO, 2000b; WHO, 1995). Table 6 shows where the diseases are endemic, the estimated number of infected people, and the sources of infection.
Most of these infections occur in Asia as a direct result of the consumption of raw or improperly cooked fish and crustaceans containing a viable and infective stage of the parasite. People’s eating habits are associated with cultural and social practices and, therefore, extremely difficult to change. Moreover, most FBT infections occur in areas where there is poverty, pollution and increasing population growth. The use of water containing human and animal faeces to fertilize plants and to feed fish, allows the life cycle of these parasites to be completed and perpetuates the infection.
As mentioned above, detection and identification of Opisthorchis, Clonorchis and Paragonium are usually based on the microscopical examination of clinical specimens by expert personnel to detect the eggs (Sukontason et al., 1999). However, the eggs of Opisthorchis are practically identical to those of Clonorchis. Slemenda et al. (1988) and Kim (1998) used specific immunological methods to diagnose paragonimiasis and clonorchiasis respectively. However, these methods are only useful to detect infected patients and are obviously dependant on the quality on the antigen preparation. One could expect variable results depending on the quality of the batch used for the analysis.
Emigration and international trade have the potential to exacerbate the problem of FBT infections. Emigrants usually take their eating habits and preferences with them, and increased international trade and improvement in communications have allowed the transport of fresh fish and crabs from one part of the world to another in a matter of hours. For example, within the EU the consumption of improperly cooked freshwater fish has traditionally been rather limited. Nowadays, however, fresh fish from Asia, Africa and South America is more easily available. The increased access of the ethnic communities in the EU to their traditional food items as well as the fact that many of those habits have become fashionable (Chinese food, sushi and sashimi) have increased the risk of FBT in Europe.
Another factor to keep in mind with respect to the possible future impact of parasitic diseases in developed countries is the dominance of the production of aquacultured species by developing countries. Howgate (1998) estimated that about 63 percent of total aquaculture production and 90 percent of fish production comes from fresh water, and implied that the greatest cause for concern in this industry - regarding food safety - is the high incidence of trematode parasites in tropical and subtropical zones. According to Brown (2000), about 85 percent percent of the fish farmed in the year 2000 came from developing countries: almost 68 percent from China, followed by India with a 6.45 percent. Other developing countries with increasing aquaculture activities are Bangladesh, Indonesia, and Thailand. Among industrial countries, Japan (2.58 percent), the United States (1.45 percent) and Norway (1.29 percent) are the leaders. China produces fish primarily in ponds, lakes, reservoirs, and rice paddies. Some 5 million hectares of land are devoted exclusively to fish farming, much of it to carp polyculture. In addition, 1.7 million hectares of rice land is used to produce rice and fish together. China breeds four types of carp that feed at different levels of the food chain. Silver carp and bighead carp are filter feeders; the grass carp feeds largely on vegetation, while the common carp is a bottom feeder, living on detritus that settles to the bottom. Most of China’s aquaculture is integrated with agriculture, enabling farmers to use agricultural wastes, such as pig manure, to fertilize ponds, thus stimulating the growth of plankton. This type of fish polyculture typically boosts the fish yield per hectare over that of monocultures by at least half, and also dominates fish farming in India, the second major producer (Brown, 2000). In order to cut the life-cycle of the trematodes this type of fish polyculture also demands the eradication of parasites from the land animals whose manure will be used to enrich the pond.
TABLE 6
Location and estimates for some endemic fish-borne trematode infections (FBT) (WHO, 1995)
|
Disease |
Caused by |
Endemic in |
Estimated infected |
Acquired from |
|
Clonorchiasis |
Clonorchis sinensis |
China |
7 million |
113 fish ssp, mainly Cypriniade |
|
Republic of Korea |
|
|
||
|
Japan |
|
|
||
|
Hong Kong |
|
|
||
|
Viet Nam |
|
|
||
|
Russian Federation |
|
|
||
|
Opisthorchiasis |
Opisthorchis felineus |
Kazakhstan |
10 million |
Cyprinidae |
|
Russian Federation |
7 million in Thailand alone |
|
||
|
Ukraine |
|
|
||
|
Thailand |
|
|
||
|
O. viverrini |
Lao People’s Democratic Republic |
|
|
|
|
Paragonimiasis |
Paragonimus westermani |
At least 20 countries. In Asia endemic in regions of China, Republic
of Korea |
22 million |
Crabs and crayfish |
Confirming the 1995 report of the World Health Organization (WHO, 1995) we have not been able to find literature concerning fast, easy methods to detect the presence of trematodes in fish or crab samples. Similarly, there seems to be a lack of data about the survival of these parasites in foodstuffs submitted to different processing conditions and how the conditions that might inactivate or kill them would affect the organoleptic properties of the fish and therefore its acceptance by the consumers (WHO, 1995).
In summary, to eradicate FBT infections there is a need to obtain information on the epidemiology of these diseases and determine to what extent aquaculture practices may enhance the problem. This should be followed by mapping the resistance of metacercaria cysts in fish (crab) flesh to relevant processing conditions and developing sustainable, effective management methods, including rural sanitation and awareness of the problem. This all requires the development of fast, uncomplicated methods for the detection and identification of the parasites in fish, snails, faeces, and environmental samples.
With the increase in the field of genomics and proteomics during the last few years, it could be reasonable to expect that parasites will be targeted as relevant organisms to map. Large scale genome projects are well advanced for the six human parasites targeted by the World Health Organization: Trypanosoma brucei, T. cruzi, Leishmania spp., schistosomes, filariae and malaria parasites (Brindley, 2000). Unfortunately, none of the FBT previously mentioned were given priority and only some sequences corresponding to P. westermanii have been published (Brindley, 2000).
It is likely that the increased trade of potentially infected fish and crustaceans into more stringent markets (European Union, United States, Japan), will force the control agencies to develop techniques for the fast and reliable identification and elimination of these parasites in food samples. The current identification techniques are fastidious and researchers involved in phylogenetic studies will require the development of straightforward techniques for their identification - including phenotypic and sequencing data. These techniques will have the potential to detect the targeted parasites in the tissues of snails, fish and crabs (see Andrews and Chilton, 2000). To illustrate the need for reliable data in parasite systematics and identification, Blair et al., (1997) found that differences in the sequences of the cytochrome c oxidase subunit I (COI) of geographically-separated populations of P. westermani were so large that they approached those seen between different species. At present, unfortunately, no such method seems to be available.
![]()
![]()
![]()