![]()
![]()
![]()
by
H. L. GOLTERMAN
Hydrobiological Institute
Nieuwersluis, The Netherlands
Abstract
Systems in which mud influences the chemistry of water are described. The rapid disappearance of phosphates from water after fertilizing can be caused by:
the iron-sulphur-phosphorus system
the calcium-carbonate-phosphorus system
the uptake by bacteria and algae (polyphosphates).
The first system is very sensitive to the redox-potential in the water or in the mud itself. Therefore the importance of measuring the oxygen demand of the mud is stressed as this process partly determines whether the mud can become anaerobic below the surface layer or not. The systems 1) and 2) are discussed in detail and are summarized. Other sorption processes, like the iron-humus-phosphorus system and the iron- or aluminium-silicon-humus systems, are also discussed. The need for much more experimental work concerning the role of these systems is stressed.
Experiments on the exchange of radiophosphorus between mud and water are described. It is necessary to distinguish between exchange and sorption processes, as has been done, for example, by Olsen. The radiophosphorus experiments often point to a high velocity of the exchange process, but there are few experiments, if any, in which the chemical nature of the active substance in the mud is well understood.
The sorption of potassium on mud is discussed. A study of the literature yielded no experimental proof of this process.
Little experimental work has been done on the mineralization of nitrogen and silicon from mud. As far as nitrogen mineralization is concerned, the only function of mud seems to be the breakdown of small quantities of not easily mineralizing substances. Perhaps it is more important that mud can convert nitrate to ammonia, even in the presence of oxygen up to 0.4 mg per litre water.
The mineralization of silicon from diatoms probably takes place completely in the mud. Nothing is known of the determining factors.
INFLUENCE DU SOL SUR LA COMPOSITION CHIMIQUE DE L'EAU ET SES EFFETS SUR LA PRODUCTIVITE
Résumé
Cette communication décrit les systèmes dans lesquels la vase a une influence sur la composition chimique de l'eau. La disparition rapide des phosphates après épandage d'engrais dans l'eau peut avoir plusieurs origines:
Le système fer-soufre-phosphore
Le système calcium-carbonate-phosphore
L'absorption par les bactéries et les algues (polyphosphates).
Le premier système est extrêmement sensible au potential redox de l'eau ou de la vase elle-même. Il faut donc accentuer l'importance de mesurer la consommation d'oxygène de la vase, car c'est en partie ce processus qui détermine si la vase peut devenir anaérobie audessous de la couche superficielle. Les systèmes 1) et 2) sont examinés en détail et résumés. D'autres processus de sorption, comme le système fer-humus-phosphore et le système fer-ou aluminium-silice-humus, sont également étudiés. L'auteur insiste sur la nécessité de poursuivre les travaux expérimentaux concernant le rôle de ces systèmes.
Il décrit des expériences sur les échanges de phosphore radio-actif entre la vase et l'eau. Il convient de distinguer entre le processus d'échange et le processus de sorption, comme l'a fait, par exemple, Olsen. Les expériences faites avec du phosphore radio-actif indiquent souvent une grande rapidité dans les échanges, mais il n'y a pratiquement pas eu d'expériences où la nature chimique de la substance active de la vase soit connue.
La possibilité de fixation par sorption du potassium sur la vase est examinée. Aucun des travaux publiés dont l'auteur a eu connaissance ne fournit la preuve expérimentale de ce processus.
Peu d'expériences ont été faites sur la minéralisation de l'azote et de la silice contenues dans la vase. En ce qui concerne la minéralisation de l'azote, la seule fonction de la vase semble être la fragmentation des faibles quantités de substances qui ne sont pas facilement minéralisables. Il est peut-être plus important de noter que la vase peut transformer l'azote nitrique en azote ammoniacal, même en présence de quantités d'oxygène allant jusqu'à 0,4 mg par litre d'eau.
La minéralisation de la silice provenant des diatomées a probablement lieu entièrement dans la vase. On ne sait rien des facteurs qui déterminent ce processus.
INFLUENCIA DEL SUELO EN LA QUIMICA DEL AGUA EN RELACION CON LA PRODUCTIVIDAD
Extracto
Se describen formas en las que el fango influye en la composición química del agua. La rápida desparición de los fosfatos del agua después de la fertilización puede ser causada por:
El primer sistema es muy sensible al potencial redox del agua o del fango. Por esto, se acentúa la importancia de medir la exigencia de oxígeno del fango, ya que este proceso determina, en parte, si el fango puede volverse anaerobic debajo de la capa superficial o no. Se examinan tambien otros procesos de sorción como el sistema hierro-humus-fósforo y el sistema hierro- o aluminio-silicio-humus. Se pone de relieve la necesidad de realizar muchos más trabajos experimentales basados en estos sistemas.
Se describen experimentos sobre el intercambio de fósforo radioactivo entre el fango y el agua. Es preciso distinguir entre procesos de intercambio y de sorción, como ha hecho, por ejemplo, Olsen. Los experimentos con fósforo radioactivo indican frecuentemente una elevada velocidad del proceso de intercambio, pero existen pocos experimentos, si es que existen algunos, en los que se conozca la naturaleza química de la sustancia activa en el fango.
Se examina la sorción de potasio en el fango. Una investigación de las obras publicadas al respecto no dio ninguna prueba experimental de este proceso. Se han realizado pocos trabajos de tipo experimental sobre la mineralización del nitrógeno y del silicio del fango. Por lo que se refiere a la mineralización del nitrógeno, la única función del fango parece ser la descomposición de la pequeña cantidad de sustancias no fácilmente mineralizantes. Quizás es más importante el hecho de que el fango puede convertir nitrato en amoníaco incluso en presencia de oxígeno hasta 0,4 mg por litro de agua. La mineralización del silicio de las diatomeas probablemente se efectúa por completo en el fango. Nada se sabe de los factores determinantes.
For the growth of fish a combination of solar energy and inorganic nutrients in a closed system, called a “food chain”, is essential. In the primary production stage, the first link of the food chain, the nutrients, are usually dissolved in water. Further on in the food chain they may also be bound to organic substances in particulate matter. The concentrations of the dissolved nutrients result from an equilibrium between their disappearance into organisms and reappearance in the mud and gas phase.
This paper is concerned with the chemical mechanism by which the mud influences the concentrations of dissolved nutrients. Only experiments directly connected with the mechanism of exchange processes are discussed, whereas all “circumstantial evidence” is omitted. When for example a fertilizer is added to water and it leads to an increased production of fish in the next year, this experiment does not prove an adsorption of the fertilizer by the mud, nor does it give any information about the mechanism of a possible adsorption.
The elements that are supposed to be “stored” in mud are the following: Fe, P, N, Ca, K, Si, and trace elements (for the latter see Gorham and Swaine, 1965). Furthermore mud plays an important role in the generation of organic substances (especially vitamins), the O2 balance of water, the pH regulation and the sulphur cycle. In most exchanges or sorption processes two or three elements are closely connected. It is therefore not possible to discuss the different elements separately.
Nearly nothing is known about the production of organic substances (including vitamins) by the mud, except that humic substances are formed there. Current research in this field (see, for example, Povoledo, 1964; Ryhänen, 1964) has not yet produced any data relating to the subject under discussion.
As the production of vitamins probably takes place by bacterial conversions, this process may also occur in the mud, but nothing is known of the determining factors. Better knowledge of these problems might be useful for increase of production.
Much work on this system has already been done by Einsele (1936; 1937; 1938), Ohle (1937; 1938) and Mortimer (1941; 1942). When a lake stratifies and the hypolimnion becomes anaerobic (and the redox potential therefore falls) (Pearsall and Mortimer, 1939) the iron in the mud (present in ferric form) will be reduced to the ferrous form. The reducing agent may be H2S, formed by anaerobic process of sulphate reduction by anaerobic bacteria like Desulfovibrio desulfuricans:
H2SO4 + H donor → H2S + 2 H2O
According to Einsele (1936), the reduction of the Fe+++ follows the reaction
2 Fe+++ + S" → 2 Fe++ + S
The Fe++ appears in the water and must be balanced by HCO3' ions. The course of this process is unknown; the reaction
FeS + 2 CO2 + 2 H2O → Fe (HCO3)2 + H2S
proposed by Einsele (1936) and Ohle (1937) seems improbable, as can be calculated from the pK's of H2CO3 and H2S.
The reduction system has been studied by Einsele (1936), who found that the reaction is not stoechiometric. In his experiments an excess of H2S was necessary to obtain a 100 percent resolution of the iron. It is not clear why the first formed Fe++ions are not directly precipitated by the remaining H2S. FeS is a very stable, insoluble compound even under aerobic conditions. It is, however, not impossible that FeS dissolves more easily when in contact with mud (Hogendijk, unpublished) than in inorganic chemistry.
Although there are no indications in the literature, it seems worth-while to investigate whether the H donor in the first reaction can reduce the iron directly. This H donor originates from the mud or directly from the extracellular products formed during photosynthesis. As this production is beyond the scope of this review, I only mention the papers of Hellebust (1965) and the work of Fogg (1964) and his co-workers (Nalewajko, 1963; Fogg and Nalewajko, 1963). The second source of the H donor consists of organic compounds released by autolysis or breakdown of dead organisms by bacteria. Golterman (1960; 1964; 1966) has shown that during these decomposition processes many products are liberated and rapidly converted by bacteria. These processes occur in both water and mud, so that it is easily understood that reduction of sulphate takes place mainly in mud, although Sorokin (1966) has shown that it happens also in water.
The quantitative experiments on the Fe-S relationship must be repeated, especially because the calculations of Einsele (1937) are partly incorrect, due to the use of wrong (out-of-date?) dissociation constants of the H2S equilibria.
At the overturn, the dissolved ferrous iron is oxidized by O2. As the pH is usually above 6 the Fe+++ formed is precipitated as Fe(OH)3, the starting point of the second reaction. When phosphates are present these are co-precipitated, either as FePO4 or adsorbed to Fe(OH)3. The last question is not a purely academic one, as adsorbed phosphorus is much more easily exchangeable than phosphorus in FePO4. It is not known whether algae can grow on the practically insoluble FePO4. Armstrong and Harvey (1951) mentioned the growth of a P-depleted diatom on FePO4, but this growth probably followed the hydrolysis of the FePO4 that takes place in sea water, as they demonstrated by the decrease in pH from 8 to 7, where the process stopped. On the other hand Ohle (1937; 1938) has demonstrated that the phosphorus bound to Fe(OH)3 precipitate can be dissolved by H2S or H2CO3 and Ca(HCO3)2 more easily than the phosphate of FePO4. Einsele (1938) described the adsorption of phosphate onto Fe(OH)3 with the adsorption isotherm Cd = K Can, in which Cd is the phosphorus concentration in the water and Ca the amount of phosphate adsorbed to 1 mg Fe(OH)3 and n and K are constants. The amount of Fe(OH)3 in the mud is mostly unknown, which fact makes the formula practically inapplicable.
The question whether these processes are important in fish ponds, brings us to the work of Mortimer (1941; 1942). He found that a redox potential below 0.2 V (associated with an oxygen concentration of 0.1 mg/l) causes reduction of iron and also that this redox potential is present in “oxidized” mud even a few centimetres below the mud-water surface. Factors influencing the thickness of the oxidized microzone are turbulent displacement of the uppermost sediments into the overlying aerated water, as well as the reducing power of the sediment itself (and therefore probably the productivity of the whole lake or pond) (Gorham, 1958). Edwards (1958) showed that introduction of larvae of Chironomus riparius caused an increased depth of the surface layer of sludge where the redox potential is high. Hayes, Reid and Cameron (1958) presented some evidence that the real oxidized layer is only one mm thick or less, by placing iron and copper wires vertically in the mud. The break in the electrode curve at about one cm is supposed to be caused by such factors as wind induced currents and presence of sulphide, or by pushing down the redox layer by the electrode. Most of the phosphorus adsorbed by the mud appeared to be in a layer of less than one mm, when determined by radio-autograph technique.
For scientific and practical purposes we should know what happens to this adsorbed phosphate when this oxidized crust is reduced, either by action from above, due to the overlying water becoming anaerobic, or by action from underneath, when a new deposit had been placed on the crust layer and the mud itself becomes anaerobic because of the O2 demand. Furthermore we should know how far the oxidized crust inhibits all transport upwards and downwards.
In very productive waters - which most fish ponds are - there is bound to be a very rapid mineralization of the organic material together with a high uptake of O2 (see section 9 below). So it is likely that the mud is mainly in a reduced state and that a “micro-stratification” occurs. In this case the phosphorus added to a fish pond as a fertilizer is very likely to be made insoluble by the formation of FePO4.
Fair, Moore and Thomas (1941) described a loss of iron from deposits, probably by reduction of iron followed by an active transport from the mud by gas bubbles (methane or hydrogen?) and the flow of water. When there is no movement in the water the ferrous compounds are oxidized and form the layer of Fe(OH)3 as described by Mortimer (1950).
The water of fish ponds generally has a high calcium content, as lime is used widely as a fertilizer. Schäperclaus even advises adding CaCO3 or CaO to acid Ca-poor ponds to raise the pH to values of about 8 in order to increase production. However, a certain optimum calcium concentration may probably not be surpassed because of the danger of formation of insoluble compounds.
The solubility of CaCO3 is low (± 15 mg/l), but is increased in the presence of CO2 (50–60 mg/l at a CO2 concentration of 0.00032 atmo.). The solubility product is ± 1 × 10-8.
In a solution containing 1.8 milliequiv. HCO3 - ions with a pH of 8.6, which solution is in equilibrium with the CO2 in the air at 20°C [Maciolek (1954) gives the pH range for fish ponds as 6.5 – 9, with an optimum around 8] the concentration of the CO"3 is
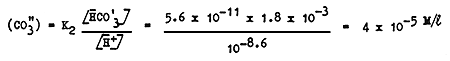
and therefore the calcium concentration is
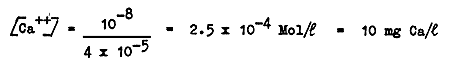
This means that the addition, in this solution, of more than 10 mg/l of calcium causes the precipitation of CaCO3.
The given example is not an unlikely one; during photosynthesis the pH may easily be higher. The figures must be recalculated for each pond after determination of its pH and HCO3ion concentration. (pH indicator papers are not reliable, as the quantity of the indicator is not small compared with the quantity of the buffering ions in water).
Up till now the precipitation of CaCO3 was not considered to be harmful. Schäperclaus even supposes that a stock of CaCO3 in the mud precipitates harmful organic acids produced by the mud. But even in the case of the very insoluble calcium oxalate this effect is not produced, as long as the calcium concentration is 10 mg/l, before the oxalate concentration surpasses 0.7 mg/l. (The solubility product of calcium oxalate is 2 × 10-9; therefore the oxalate concentration in equilibrium with a calcium concentration of
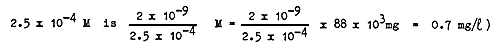
For the whole water mass this seems to be an unattainable concentration. The calcium salts of many other organic acids are even more soluble than calcium oxalate. Furthermore, no experiments could be found in the literature that proved the production of harmful organic acids by mud.
The second advantage mentioned by Schäperclaus, the buffering capacity against acid production, is again hypothetical; a saturated CaCO3 + Ca(HCO3)2 solution, without any extra solid CaCO3 is more than sufficient to buffer the quantities of acid produced by mud in a natural system. When a mud (with a metabolic rate: O2 uptake of 5 g/m2/day, see Section 9) produces 10 percent of this amount as an organic acid with 3 C atoms under anaerobic conditions, it produces

which is low compared with the amount of HCO'3 in the given example.
The addition of too much CaCO3 to a pond seems to me to be only disadvantageous because of precipitation of phosphorus.
Hepher (1958) has given considerable attention to the problem of precipitation of Ca3(PO4)2 and has given a useful diagram for calculation of the amounts of Ca and PO4 that can exist together in a solution at different pH values. His experimental results sometimes agree with the calculated values. [Hepher used 1 × 10-25 as the solubility product of Ca3(PO4)2 in his calculations, and mentioned still lower values. But even 1 × 10-25 is much lower than can be calculated from the solubility of Ca3(PO4)2 (20 – 30 mg/l), which gives 10-20 – 10-21. This enormous difference may be caused by the fact that Ca3(PO4)2 in water forms compounds such as hydroxyapatites or even their Cl' or F' salts. (See Remy, 1956, pp.624 and 638)]. However, the differences are too large to be neglected and in table 6 of his paper the possibility of precipitation of 3 Ca3(PO4)2.2Ca(OH)2 or Ca5(PO4)3OH must be considered. (See section 5 below). It might have been interesting to know the chemical composition of the precipitates occurring in his experiments, as it can be concluded that Ca is precipitated in quantities largely exceeding those of the phosphate, which leads to the hypothesis that very complicated precipitates occurred.
Hepher (1958) certainly shows a correlation between CaCO3 content of the mud and
Ca2(PO4)3 precipitation, but no proof of the proposed reaction between CaCO3 and
PO4ions. Barrett (1953) also supported the fact that the disappearance of added phosphorus
from epilimnial water is related to alkalinity. Furthermore, Hepher's idea
that no adsorption onto heavy metals can take place because of the high pH is not
necessarily true. Ohle (1937–1938) indeed demonstrated that PO"4' adsorbed onto
Fe(OH)3 could be washed out with Ca(HCO3)2 solutions. But in ion-exchange processes
the continuous renewal of the solvent shifts the equilibrium Fe(OH)3~P Fe(OH)3,
+ H3PO4 to the right, by permanently removing one of the reaction's products. Ohle's
experiment only proved that adsorption of phosphate to Fe(OH)3 is less at pH 8.6 than
at pH 6.0.
Fe(OH)3,
+ H3PO4 to the right, by permanently removing one of the reaction's products. Ohle's
experiment only proved that adsorption of phosphate to Fe(OH)3 is less at pH 8.6 than
at pH 6.0.
Hepher also considers the possibility that phosphate is taken up by the algae in excess of their normal requirements. In my opinion the presence of polyphosphates in algae and the P uptake by bacteria are more important than Hepher thinks, as can be calculated by their rapid turnover times (for algae see for example Coffin, 1949). As the polyphosphates are beyond the scope of this paper, I only mention the papers of Baker (1964), Curnutt and Schmidt (1964) and Kuhl (1962).
The conclusion about the iron-sulphur-phosphorus and the calcium-carbonate-phosphorus system must be that it is not yet possible to know which type of sorption or precipitation is the main process in a given mud after the addition of phosphate.
It is extremely urgent to know more about the systems combined. It is desirable to start quantitative investigations with the Ca-CO3-PO4-system and the F-S-PO4-system separately, and to combine both systems after we have learned to distinguish between FePO4, Fe(OH)3~P and [Ca3(PO4)2], whatever the composition of the last precipitate may be. The hypothesis (Neess, 1949) that the addition of lime before fertilization with phosphate will save the latter from precipitation in iron-phosphorus complexes, still seems very doubtful. For the study of the entire mud system radiophosphorus experiments and determinations of iron and calcium activities must be carried out simultaneously. Too little attention has been paid to the determination of organic phosphates, both dissolved and particulate, in fish ponds. Before any conclusions about the uptake of phosphorus by the mud are drawn, the turnover times of the phosphorus compounds during primary production must be measured. P32 combined with C14 may be used by all workers applying the C14 technique.
Humic acids are supposed to form complexes with heavy metals, mostly Fe, by chelation (cf.EDTA).
However, as chelation leads only to an equilibrium, the iron linkage to the humic acids in dissolved state will always be counteracted by high calcium concentration. This difficulty is overcome by the finding of Shapiro (1964), who observed that dissolved iron, which in oxygenated water must be in the ferric form, is bound to particles (of yellow acids) larger than 0.5/μ, and he described some properties of such complexes. Shapiro believes that in highly coloured waters the large quantities of iron held in apparent solution are in the form of a protected colloid and that chelation plays a relatively minor role. The bound metal in its turn can combine with phosphorus compounds.
I could find in the literature no proof of the assumption that the calcium in lime can displace other fertilizing substances from organic colloidal systems (Neess, 1949), nor of the flocculation of the colloidal humus gels and the ability of these colloids to liberate hydrogen. As long as our knowledge of the structure of the humic acids is only hypothetical, one has to be very careful in proposing chemical mechanisms. Although there appears to be a close structural relationship between the organic matter present in lacustrine and marine sediments and the waters on one hand, and that present in soils on the other hand (see for example Povoledo, 1964), it is not safe to use data about adsorption from soil chemistry, as the equilibria in the aquatic system may differ from the discontinuous soil system.
Ohle (1955; 1964) has made some investigations on the adsorption to humic acids and demonstrated a linkage of phosphorus and silicon to iron and aluminium-humic acid complexes. The objection might be made that he used “Merck” humic acids. It is preferable, although more difficult, to use “native” humic acids, to avoid the risk of a change in sorption properties by purification.
Ryhänen (1964) also described aluminium-humic acid complexes. Ohle furthermore demonstrated a linkage of silicate to ferric and aluminium hydroxide. The question, whether iron- and aluminium silicates (as kaolinite Al2[Si2O5-] (OH)4 and montmorillonite AlSi2O5(OH)) with or without iron oxide behave as ion exchangers under natural conditions, remains to be proved.
The same objection must be made to the supposition that minerals like apatite 3Ca3(PO4)2.Ca(F,Cl)2, phosphorite 3Ca3(PO4)2.Ca(OH)2, vivianite Fe3(PO4)2.8 H2O and wavellite 3Al2O3.2P2O3.12 H2O regulate the phosphorus concentration in the lake water. Whether the Ca of minerals in mud is exchangeable like the Ca in soil clay (see for example Borland and Reitemeier, 1950) must still be investigated.
In experiments with radiophosphorus always a rapid exchange between P31 and P32 can be found. This exchange which must be distinguished from adsorption processes, can give some information about the nature of phosphorus compounds present, but not about the quantity of phosphorus “stored” in mud. In literature a good distinction between the exchange process: X P31 O4 + P32O4 = X P32O4 + P31O4 and the adsorption process: X(Y)3 + PO"4' = X PO4 + 3 Y is not always made. Furthermore the fact must be mentioned that under conditions leading to the formation of phosphorite in nature, the product is not Ca3(PO4)2 but hydroxylapatite 3Ca3(PO4)2.Ca(OH)2 or Ca5(PO4)3(OH). This compound is also formed when tert. calciumphosphate is suspended in water (which compound therefore cannot be prepared in the pure state by precipitation from a solution), while even CaHPO4 is decomposed by water by the reaction:
7 CaHPO4 + H2O → Ca5(PO4)3OH + 2 Ca(H2PO4)2
For further information see Remy, 1956: pages 624, 638.
Livingstone and Boykin (1962) studied the phosphorus in the sediment of Linsley Pond and suggested that a large quantity of the phosphate is bound to the mineral material below the gyttja, by sorption reactions. The mineral part was not present as apatite, as appeared from the solubility in acids. In their interesting discussion, they remark that high phosphorus binding capacity is correlated with high mineral content of lake mud. A comparison of the adsorption capacities of the gyttja and the mineral material as a function of the phosphorus concentration of the overlying water would have been very interesting.
Macpherson, Sinclair and Hayes (1958) studied the pH dependence of phosphate sorption by dried mud of different lakes and the ash of this mud. Both curves from eight lakes studied showed a maximum sorption at pH values 5 – 7, the ash giving off more phosphate than the dried mud when shaken with distilled water. The organic part of mud appeared to moderate the phosphate release at the end of the pH range. Both facts were confirmed by shaking the mud with one ppm of phosphate. However, it may not be concluded from their figures whether they measured a pH effect (a sorption reaction) or an effect by changing the ionic strength (an exchange reaction). The activity of the ash compared with the mud as a whole may also be an activation effect by heating. Furthermore they found a similarity of the pH effect on lake mud ash, to the effects on betonite, fuller's earth and ferric hydroxide as described in the literature.
The distribution of p32 between mud and water was studied by Hayes and Phillips (1958), who found in the first place that in natural Jenkin sampler cores, in artificial cores and in bottles in which dredged surface mud was packed by centrifuge, the phosphorus equilibration pattern and rate were the same, which proved that layering was relatively unimportant. Furthermore they found that in the presence of bacteria the amount of P32 remaining in the water at equilibrium was greater than where antibiotic had been added. Beside the two explanations Hayes and Phillips have given, it is also possible - considering the chemical formulae of their antibiotics - to propose the hypothesis that the antibiotics themselves influenced the sorption system.
They found turnover times to be: for bacteria or phytoplankton ± five minutes, confirming the results of Coffin (1949), for the water of the whole lake one week, for mud in a bottle half a week (without bacteria two weeks), and for lake sediments in nature (including rooted aquatics) one month. Zooplankton was unable to use phosphorus until bacteria had made it organic; its turnover time was than one day.
Pomeroy (1965) studied the phosphorus exchange of P32 with cores and suspended sediments containing as principal clay minerals kaolinite and montmorillonite. They found a two-step ion exchange between the clay minerals and the phosphate of water, plus an exchange between micro-organisms and water. The latter appeared to be trivial in undisturbed sediments but to equal the inorganic process in suspended sediments. They do not mention whether the sediments contained calcium carbonate or calcium phosphate precipitates.
Although findings may vary from lake to lake, these results may be of great help in a clearer understanding of the phosphorus balance in fish ponds.
Radiophosphorus experiments in lake sediments were carried out by Olsen (1958; 1964), who made the right distinction between exchange and adsorption, but whose paper did not receive the attention it deserved. Olsen used a coarse sediment from 3 metres and a finely grained deep water sediment, both in reduced as well as oxidized state. He found that the relations between the phosphorus in the water and that in the sediment could be described by the same mathematical formula, with reservation in respect to the phosphate release from the reduced sediments, the relation being a hyperbolalike function of the form
b = Kb · C-Vb
in which b is the exchange phosphorus in γ's P per g dry matter while Kb and Vb are constants and C is the concentration in the solution in γ's P per ml.
Addition of this exchange quantity to the net adsorbed quantity (a) gives the gross adsorbed quantity (A), which Olsen described by the Freundlich adsorption isotherm
A = K.CV
The full mathematical description of the direct measurable net adsorbed quantity was calculated by Olsen as
a = A - b = K. CV -Kb .C-Vb
It is shown by Olsen that the exchange of the phosphorus is a very rapid process and that uptake of phosphorus from water by algae will be followed by a release of phosphorus from the sediments, with different constants for oxidized or reduced sediments. In the oxidized state sediment exchanges and adsorbs more phosphorus than in the reduced state, which is another argument for investigating the extent of reduction of fish pond bottoms. In the reduced state the sediment from deeper water showed no adsorption of phosphorus - and therefore only release - when the concentration of phosphorus was below 2 mg P/l, which is a high value.
It would be interesting to know whether the divergent fate of phosphorus in reduced “deep” sediments is related to redox-potentials and iron metabolism. It can be imagined that the reduced “deep” sediment has lost all its active iron and that the adsorption of phosphorus from solutions containing more than 2 mg P/l takes place by a different process, for example precipitation with calcium.
Schäperclaus mentioned in his review that K is adsorbed to the mud during winter and released during summer. He quotes a study of Breest (1924) as the only source of this information. Neess (1949) again quotes Breest about evidence for K exchange between mud and water. Breest's paper, however, does not give the information that Schäperclaus and Neess report. In the first place Breest did not measure potassium sorption but sorption of Cl'ions which need not be the same as K sorption. Doubtless mud contains a large reservoir of potassium, but as the K-ion has only one electrovalence and KCl in a solution is completely ionized, the binding of the K to the mud leaves the Cl'ion in the water. Moreover his paper, dealing also with the flocculation of colloids, lacks technical information, for example about the influence of time on his results.
Edwards and Rolley (1965) studied the exchange between interstitial water and water overlying mud deposits. From their figures it appears that no adsorption of Li takes place. When the K linkage to the mud follows an ion exchange pattern it seems likely that lithium, too, should have been adsorbed. Their experiments, therefore, give “circumstantial evidence”, but no proof, of a non-ionic nature of the linkage.
Wiklander (1950) wanted to remove K from a micaceous clay and needed therefore 21 days of treatment with N solutions of HCl, NaCl, NH4Cl or CaCl2 at a temperature of 65 to 70°C. Although this clay came from soil, this fact shows that the K fixed in minerals is not always easily exchangeable.
The role of nitrogen in fish ponds has been reviewed by Hepher (1952b) and Neess (1949). There is no general answer to the question whether N-manuring is necessary because the ponds are so very different. However, the fact must be stressed that elimination of one limiting factor (e.g. phosphorus) automatically causes a second factor to become limiting (probably nitrogen, sometimes perhaps silicon or iron).
For a good growth of algae the quantities of nitrogen and phosphorus (in mg) should be available in a ratio varying from 10:1 to 4:1, because the same ratio normally occurs within the algae. (When polyphosphates are stocked in the algae, the lower ratio will be approached).
By determining the turnover time of phosphorus in the algae, the amount of nitrogen required can be estimated. However, only part of this amount has to be generated by mud, as nitrogen is rapidly remineralized after the death of the algae (Golterman, 1960). In our laboratory experiments we found that after three to five days, 50 – 75 percent of the incorporated nitrogen is mineralized to ammonia by bacteria. As the sinking rate of plankton is in the order of magnitude of one metre a day in stagnant water, it is most likely that in fish ponds which are shallow and strongly influenced by wind the algae will probably be broken down before they reach the mud. In an experimental plastic pond without mud at the bottom we found a 100 percent mineralization in two to three days after a bloom of Chlamydomonas had died off. Ohle (1962) has also described a 90 percent mineralization of the incorporated nitrogen, in the upper layer of a lake, in a few weeks. During this process the algal material is converted to ammonia and CO2, bacterial substance and soluble organic substances. The ratio of the products formed is determined by the C/N ratio of the plankton, which can be estimated as about C5H7NO2 (Busch, 1965). The remaining, not easily mineralizing compounds must be broken down in the mud, probably also by bacteria. A study of this process was made by Fair, Moore and Thomas (1941b), whose work on O2 uptake will be discussed later. They found that the quantities of soluble nitrogen products formed are determined by physical conditions of environment, relative activity of fermentation, and variations in the initial concentrations and constitution of nitrogenous matter in the deposits. During their experiments (differing in time from 145–450 days) one quarter to three quarters of the total nitrogen in the mud was observed to remain in the deposits. Schäperclaus thinks that a neutral mud stimulates this process (an argument in favour of Ca -manuring) but this theory lacks experimental proof. Very little is known of the factors stimulating the mineralization process.
An important factor to determine which substances are released is the redox potential. Kusnetzow and Musnetzowa (1935), (see Mortimer, 1942, p. 199) found for example that the reduction of formic acid to methane took place most actively in mud at a redox potential of E7 = - 0.12 V., with C and N supply as the controlling factors.
There are two more processes by which mud can influence the N cycle:
Adsorption of NH+4 to the colloidal parts. No experimental proof of this process is found in the literature.
Reduction of nitrate to ammonia. Mortimer (1941) has found that below a redox potential of E7 = + 0.35 V. only ammonia can be detected (this potential was associated in his work with an oxygen concentration of 0.4 mg/l).
Owens and Edwards (1963) have shown that mud, even when the overlying water contains O2, reduces nitrate, principally to nitrogen or ammonia. The nitrate acts as an alternative hydrogen acceptor to the oxygen. Edwards and Rolley (1965) found that nitrate reduction by mud is high when either the O2 consumption by the mud or the nitrate concentration in the water is high.
From their findings it may be concluded that in fish ponds, where a high NO'3 concentration is desirable, the nitrate will easily be reduced to ammonia when the mud has a high O2 consumption, probably causing a harmful effect on the fish.
If any exchange processes of silicon compounds take place in the mud, they must have the character of ion exchange processes. It has been found that the silicates in the hypolimnion of stratified lakes is constantly increasing. It is not known whether this increase is directly influenced by the fall of the redox potential. Of course silicates themselves cannot be reduced, but it might be interesting to find out whether the rise in iron or calcium concentration leads to a higher silicate concentration in the water. The nature of the silicon compounds in the water is not yet known, nor do we know which compounds are determined by the normal silicon-molybdate complex reagent.
Part of the silicon in the water originates from dead diatoms. This mineralization is a non-enzymatic slow process. Jørgensen (1955) found for example that for Nitzschia linearis at pH = 10 a maximal amount of 20 percent of the silicon dissolved after 85 days, whereas at the same pH for Thalassiosira nana this amounted to 97 percent after 37 days. These differences may be explained by the assumption that there exists “functional” and “stored” silicon, with different mineralization times (Golterman, 1960). We found that the mineralization of the silicon of Stephanodiscus hantzschii depends on the bicarbonate concentration of the suspending medium and on the concentration of the dissolved silicate.
Mud always takes up oxygen for chemical and bacteriological decomposition processes. By the addition of poisons like HgCl2 or toluene a distinction between these processes can be made, but this has probably no practical significance for fish ponds. The extent of the uptake, however, strongly influences the O2 concentration in the water and thereby many redox reactions. The measuring technique of Knowles, Edwards and Briggs (1962) appears to be suitable even when no sophisticated laboratory is available. Edwards recently measured an uptake of 0.1 and 0.2 g O2/m2/h, which means 2.4 – 4.8 g/m2/day. When a water layer of one m is assumed, this means that 50 percent of the dissolved O2 is consumed by the mud. They found no significant change in O2 consumption with mud depth ranging between 4 and 17 cm, contrary to the findings of Fair, Moore and Thomas (1941 a,b,c) and Baity (1938). The latter found that the relation between O2 demand (Y) and sludge depth (X) is
Y = 2700 X0.485
Probably in each case the composition of the mud and also the accumulation time were different.
Edwards (1958) stressed the importance of invertebrates in affecting the physical, chemical and microbiological conditions within the deposits when the benthal O2 demands are measured.
Fair, Moore and Thomas (1941 a,b,c) studied some other factors influencing the O2 uptake of river mud and have given some mathematical formulae concerning the effect of temperature. Furthermore they studied (1941 a and c) the decrease of the potential oxygen demand of the decomposable matter, caused by anaerobic decomposition. This decomposition gains in importance when the sediments grow in depth, although the aerobic processes remain the most important quantitatively. Although their work on river muds is connected with sewage and sludge purification we must keep in mind that comparable processes are bound to occur at the bottom of the always very eutrophic fish ponds.
Armstrong, F.A.J. and H.W. Harvey, 1951 The cycle of phosphorus in the waters of the English Channel. J.Mar.biol.Ass.N.S., 29:145–62
Baity, H.G., 1938 Some factors affecting the aerobic decomposition of sewage sludge deposits. Sewage Works J., 10:539–68
Baker, A.L., 1964 Polyphosphate metabolism during nuclear division in synchronous growing Chlorella. Biochim.biophys.Acta, 82(3):624
Barret, P.H., 1953 Relationships between alkanity and adsorption and regeneration of added phosphorus in fertilized trout lakes. Trans.Amer.Fish.Soc., 82:78–90
Borland, J.W. and R.F. Reitemeier, 1950 Kinetic exchange studies on clays with radioactive calcium. Soil Sci., 69:251–60
Breest, F., 1924 Uber die Beziehungen zwischen Teichwasser, Teichschlamm und Teichuntergrund. Arch.Hydrobiol., 15:422
Busch, A.W., 1965 Determination of biologically degradable carbon concentration in water. Hydrobiologia, 26(1–2):128–9 Symposium on new methods of determining biogenic and organic substances in water and organisms; held in 1963 in Zivohośt, Czechoslovakia.
Coffin, C.C. et al., 1949 Exchange of materials in a lake as studied by the addition of radioactive phosphorus. Canad.J.Res.(D), 27:207–22
Curnutt, S.G. and R.R. Schmidt, 1964 Possible mechanisms controlling the intracellular level of the inorganic polyphosphate during synchronous growth of Chlorella pyrenoidosa. Biochim.biophys.Acta, 86(1):201
Dussart, B., 1966 Limnologis; l'étude des eaux continentales. Paris, Gauthiers-Villars, 618 p.
Edwards, R.W., 1958 The effect of larvae of Chironomus riparius Meigen on the redox potentials of settled activated sludge. Ann.appl.Biol., 46(3):457–64
Edwards, R.W. and H.L.J. Rolley, 1965 Oxygen consumption of river muds. J.Ecol., 53(1):1–19
Einsele, W., 1936 Uber die Beziehungen des Eisenkreislaufs zum Phosphatkreislauf im eutrophen See. Arch.Hydrobiol., 29:664–86
Einsele, W., 1937 Physikalisch-chemische Betrachtung einiger Probleme des limnischen Manganund Eisenkreislaufs. Verh.int.Ver.Limnol., 5(3):69–84
Einsele, W., 1938 Uber chemische und kolloidchemische Vorgänge in Eisen-Phosphat Systemen unter limnochemischen und limnogeologischen Gesichtspunkten. Arch.Hydrobiol., 33:361–87
Fair, G.M., E.W. Moore and H.A. Thomas, 1941a The natural purification of river muds and pollutional sediments. Sewage Works J., 13(2):270–307
Fair, G.M., 1941b The natural purification of river muds and pollutional sediments. Sewage Works J., 13(4):756–79
Fair, G.M., 1941c The natural purification of river muds and pollutional sediments. Sewage Works J., 13(6):1209–28
Fogg, G.E., 1964 Glycollic acid as an extracellular product of phytoplankton. Chem.Weekblad, 60(38):515–9
Fogg, G.E. and C. Nalewajko, 1963 Experiments concerning the extent of liberation of extracellular products by plankton algae. Proc.roy.Soc.(B), 157:381–2
Golterman, H.L., 1960 Studies on the cycle of elements in fresh water. Acta bot.neerl., 9:1–58
Golterman, H.L., 1964 Mineralization of algae under sterile conditions or by bacterial breakdown. Verh.int.Ver.Limnol., 15:544–8
Golterman, H.L., Mitt.int.Ver.Limnol., 14 (in press)
Gorham, E., 1958 Observations on the formation and breakdown of the oxidized microzone at the mud surface in lakes. Limnol.Oceanogr., 3(3):291–8
Gorham, E. and D.J. Swaine, 1965 The influence of oxidizing and reducing conditons upon the distribution of some elements in lake sediments. Limnol.Oceanogr., 10(2):268–79
Hayes, F.R. and J.E. Phillips, 1958 Lake water and sediment. 4. Radiophosphorus equilibrium with mud, plants and bacteria under oxidized and reduced conditions. Limnol. Oceanogr., 3(4):459–75
Hayes, F.R., B.L. Reid and M.L. Cameron, 1958 Lake water and sediment. 2. Oxidation-reduction relations at the mud-water interface. Limnol.Oceanogr., 3(3):308–17
Hayes, F.R. et al., 1952 On the kinetics of P exchange in lakes. J.Ecol., 40:202
Hellebust, J.A., 1965 Excretion of some organic compounds by marine phytoplankton. Limnol. Oceanogr., 10(2):192–206
Hepher, B., 1952a The fertilization of fish ponds. 1. Phosphates. Bamidgeh, 4(718):131–4
Hepher, B., 1952b The fertilization of fish ponds. 2. Nitrogen. Bamidgeh, 4(10/11/12):220–3
Hepher, B., 1953 The fertilization of fish ponds. 3. Organic Matter. Bamidgeh, 5(1/2):19–24
Hepher, B., 1958 On the dynamics of phosphorus added to fish ponds in Israel. Limnol. Oceanogr., 3:84–100
Jørgensen, E.G., 1955 Solubility of the silica in diatoms. Physiol.Plant., 8:846–51
Kanai, R., Sh. Miyachi and Shigetoh Miyachi, 1963 Light-induced formation and mobilization of polyphosphate “C” in Chlorella cells. In Studies on microalgae and photosynthetic bacteria, ed. by Japanese Society of Plant Physiologists, Tokyo, pp. 613–8
Knowles, G., R.W. Edwards and R. Briggs, 1962 Polarographic measurement of the rate of respiration of natural sediments. Limnol.Oceanogr., 7(4):481–3
Kuhl, A., 1962 Inorganic phosphorus uptake and metabolism. 5. Condensed inorganic phosphates. In Physiology and biochemistry of algae ed. by R.A. Lewin, New York, Academic Press, pp. 218–29
Kusnezow, S.I. and Z.I. Kusnezowa, 1935 Die bakteriologische und die chemische Untersuchung von Seeschlammen im Zusammenhang mit der Gasausscheidung aus dem Seegrund. Trudy Limnol.Sta.Kosíne, 19:127–44 (Only in Russian)
Lewis, G.C., G.O. Baker and R.S. Synder, 1950 Phosphate fixation in calcareous soils. Soil Sci., 69:55–62
Livingstone, D.A. and J.C. Boykin, 1962 Vertical distribution of phosphorus in Linsley Pond mud. Limnol.Oceanogr., 7(1):57–62
Maciolek, A., 1954 Artificial fertilization of lakes and ponds. Spec.sci.Rep.U.S.Fish Wildl.Serv.-Fish., (113)
Macpherson, L.B., N.R. Sinclair and F.R. Hayes, 1958 Lake water and sediments. 3. The effect of pH on the partition of inorganic phosphates between water and oxidized mud or its ash. Limnol.Oceanogr., 3(3):318–26
Mortimer, C.H., 1941 The exchange of dissolved substances between mud and water in lakes. J.Ecol., 29:280–329
Mortimer, C.H., 1942 The exchange of dissolved substances between mud and water in lakes. J.Ecol., 30:147–201
Mortimer, C.H., 1950 Underwater “soils”; A review of lake sediments. J.Soil Sci., 1:63–73
Mortimer, C.H. and C.H. Hickling, 1954 Fertilizers in fish ponds. Fish.Publ.,Lond., (5):155 p.
Nalewajko, C., N. Chowdhuri and G.E. Fogg, 1963 Excretion of glycollic acid and the growth of a planktonic Chlorella. Microalgae a.Photosynthetic Bact., 171–83
Neess, J.C., 1949 Development and status of pond fertilization in Central Europe. Trans. Amer.Fish.Soc., 76:335–58
Ohle, W., 1937 Kolloidgele als Nährstoffregulatoren der Gewässer. Naturwissenschaften, 25 (29):471–4
Ohle, W., 1938 Die Bedeutung der Austauschvorgänge zwischen Schlamm und Wasser für den Stoffkreislauf der Gewässer. Vom Wasser, 13:87–97
Ohle, W., 1955 Ionenaustausch der Gewässersedimente. Mem.Ist.ital.Idrobiol., Suppl. (8):221–45
Ohle, W., 1962 Der Stoffhaushalt der Seen als Grundlage einer allgemeinen Stoffwechsel-dynamik der Gewässer. Kieler Meeresforsch., 18(3):107–20
Ohle, W., 1964 Kolloidkomplexe als Kationen- und Anionenaustauscher in Binnengewässern. Vom Wasser, 30:50–64
Olsen, S., 1958 Phosphate adsorption and isotopic exchange in lake muds; experiments with P 32. Preliminary rep. Verh.int.Ver.Limnol., 13:915–22
Olsen, S., 1964 Phosphate equilibrium between reduced sediments and water: Laboratory experiments with radioactive phosphorus. Verh.int.Ver.Limnol., 15:333–41
Owens, M. and R.W. Edwards, 1963 Oxygen studies. Proc.Soc.Wat.Treatm.Exam., 12:126–45
Pearsall, W.H. and C.H. Mortimer, 1939 Oxidation-reduction potentials in waterlogged soils, natural waters and muds. J.Ecol., 27:483–501
Perkins, A., 1947 Phosphate solubility in relation to cations and pH: Magnesium. Proc.Soil Sci. Soc.Amer., 12:185–7
Pomeroy, L.R., E.E. Smith and C.M. Grant, 1965 The exchange of phosphate between estuarine water and sediments. Limnol.Oceanogr., 10(2):167–72
Povoledo, D., 1964 Some comparative, physical and chemical studies on soil and lacutrine organic matter. Mem.Ist.ital.Idrobiol., 17:21–32
Remy, A., 1956 Treatise on inorganic chemistry. 1. Amsterdam, London, Elsevier P., pp. 624–5,638
Ryhänen, R., 1964 Beobachtungen über “Humuskolloide” in angewandter Limnologie. Verh.int. Ver.Limnol., 15:276–83
Schäperclaus, W., 1961 Lehrbuch der Teichwirtschaft; zweite erw.Auflage. Berlin u. Hamburg, P.Parey, 582 p.
Shapiro, J., 1964 Effects of yellow organic acids on iron and other metals in water. J.Amer. Wat.Wks.Ass., 56(8):1062–82
Sorokin, Yu. I., 1966 Proceedings of the Symposium, in Pallanza, on the Primary Productivity measurements Mem.Ist.ital.Idrobiol., 18(Suppl.):187–205
Wiklander, L., 1950 Fixation of potassium by clays saturated with different cations. Soil Sci., 69:261–80
![]()
![]()
![]()