![]()
![]()
![]()
In the last few years embryo transfer has developed into an important biotechnological approach to cattle breeding. Apart from being used as a means of amplifying the number of genetically valuable dams by increasing the number of offspring, the technique has also been used for preserving genomes.
Techniques allowing collection and transfer of cattle embryos have been simplified considerably by the development of the non-surgical techniques. Nevertheless embryo transfer programmes are still hampered by pronounced differences in the reactions of individual donor animals towards superovulation. This is important because the availability of a large number of cattle embryos capable of further development is an important prerequisite for further success.
At the time of birth ovaries of female mammals contain thousands of oocytes. Only a few are actually utilized for active reproduction. In cattle the simultaneous collection of a large number of oocytes can be achieved by superovulation which is induced by treatment with gonadotropic substances. These hormones overstimulate the ovaries and hence induce multiple ovulations.
The most widely used gonadotropic hormones capable of inducing superovulation in cattle are pregnant mare's serum gonadotropin (PMSG) and porcine follicle stimulating hormone (pFSH) which is obtained from porcine pituitaries. Another hormone which is used in human medicine is human menopausal gonadotropin (HMG). Since PMSG has a long biological half-life, a single injection is sufficient for treatment, while FSH which has a biological half-life of five hours only has to be administered for four to five days twice daily in decreasing doses in order to stimulate ovaries sufficiently.
The problem of unsatisfactory reactions towards superovulation treatment has been recognized ever since Casida et al. (1943) described the first successful attempts by treating cattle between the 16th and 18th day of the oestrous cycle with pituitary extracts obtained from cattle and sheep. They noted that approximately 30 percent of the animals treated did not display enhanced development of follicles.
Schilling and Holm (1963) have injected PMSG twice at the onset and at the end of the luteal phase to elicit superovulation. This treatment also did not reduce significantly individual variability.
Pretreatment of animals with progesterone or oestrogens followed by administration of PMSG or FSH has been used by several authors. Rowson (1951) has described lower ovulation rates when animals were stimulated with PMSG in the follicle rather than the luteal phase.
The availability of luteolytic substances such as prostaglandins and their analogues has facilitated the development of regimes allowing controlled termination of ovulation. The problem of oestrous induction has been solved in this way since a combined treatment with PMSG and PGF 2α allows successful superovulation between day 5 and 16 of the oestrous cycle. The first successful treatments have been described by Elsden et al. (1974), Rasbech (1974), Sreenan and Beehan (1974), Mickelsen (1974), and Moore (1975) who administered prostaglandin or one of its analogues by the intramuscular or intrauterine route. Doses were given either once, 24 hrs after injection of PMSG, or twice, 24 and 48 hrs after injection of PMSG.
Apart from luteolytic drugs gestagens are also suitable for combined treatment in conjunction with PMSG (Figures 2,3). Gestagens can be used at any stage of the oestrous cycle to induce ovulation since, due to the rebound effect, the animals come on heat shortly after hormone treatment is discontinued. Suitable forms of presentation are the vaginal pessary PRID™ or the ear implant NORGESTOMET™. These repository preparations can be employed in a much more controlled way than the orally administered gestagen preparations used formerly.
Although PMSG and FSH stimulate the ovaries to the same extent, it is predominantly pFSH that is used in the US and Europe.
Embryos are obtained on day 7 from the uterus by a non-surgical technique after migration through the oviduct has been completed (at day 5 of the oestrous cycle). This is usually achieved by aspirating the cervical mucus and inserting a catheter with an insemination pipette into the uterus. The catheter is fixed cranially to the bifurcation in one of the uterine horns by using a small balloon (Figure 4). Medium (Dulbecco's phosphate-buffered saline) is then introduced into the uterus to resuspend the embryos, which can then be collected by drainage. Several different catheter techniques for collecting embryos have been described:
Use of a two-way catheter with metal drainage hole (Lampeter 1977 a, b);
Use of a two-way catheter with rubber drainage hole (Baumgärtner et al., 1977);
Use of a rubber three-way catheter (Brand et al., 1978);
Use of a metal three-way catheter (Sugie, 1965).
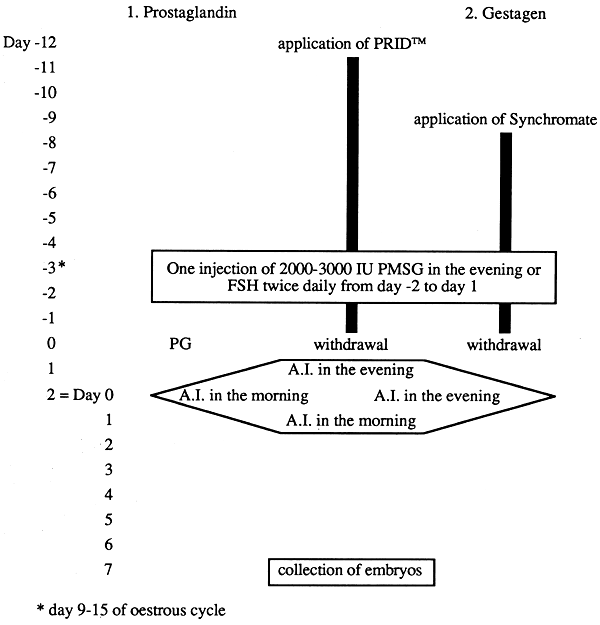
Figure 2: Time Schedules for Superovulation
The medium can be processed in different ways to collect the embryos:
Infusion of the medium by exploiting natural gravity: the uterus can be flooded with up to one litre of fluid by using a suitable tubing system. The use of a three-way catheter allows the fluid to be drained, again by gravity, immediately after the washing process (flow-through technique). The use of two-way catheters usually requires a T-valve. After a certain amount of fluid has been infused the contents of the uterus are syphoned off (closed system technique).
Infusion of the medium by means of a syringe: Upon infusion of a certain amount of fluid, usually 50–80 ml, the fluid is immediately removed by suction. This technique is usually employed in conjunction with two-way catheters.
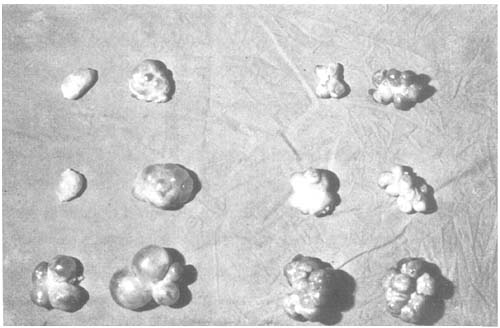
Figure 3: Pairs of Ovaries of 6 Superovulated Cows Without (Left) and With (Right) Application of PMSG Antiserum
Several different techniques have also been described for processing the recovered medium:
Depending upon the amounts of liquid the recovered fluids are kept in one or several 500 ml graduated cylinders for approximately 20 min to let the embryos settle at the bottom. The supernatant is either carefully decanted or syphoned off through a filter system (Figure 5). The latter technique has the advantage of reducing waiting times significantly.
The recovered medium is put into funnel-shaped beakers. This technique too is based on the fact that embryos are denser than the medium and therefore collect at the tip of the beaker. The supernatant is decanted.
The medium is filtered after recovery from the uterus. The filter system may be integrated into the outlet of the flushing system. Alternatively the embryos are separated from the fluid after it has been drained from the uterus by a suitable filter system.
a) 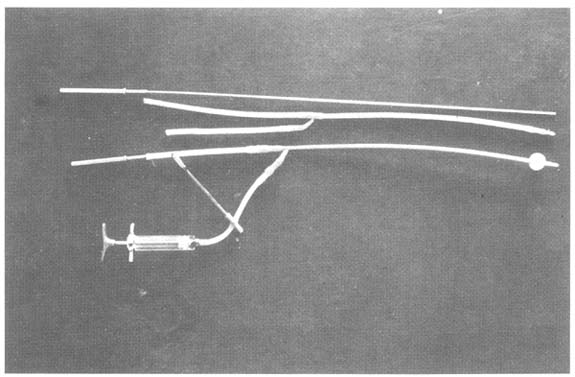
b) 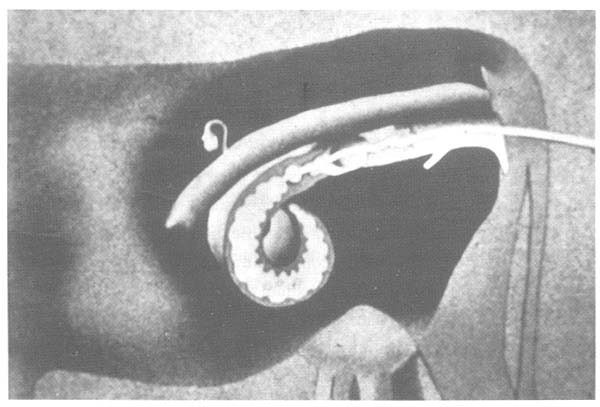
Figure 4: Tools Required for Non-Invasive Collection of Embryos (a), and Schematical Description of the Process (b)
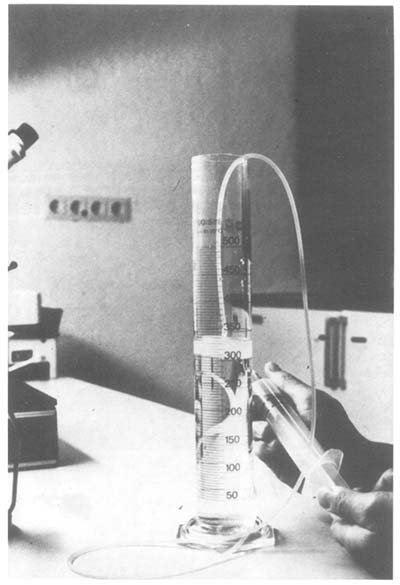
Figure 5: Syphoning off Medium to Reduce the Amount of Liquid Before Embryos are Collected
The concentrated medium (see items 1,2) is then put into one or several large Petri dishes for further microscopic inspection. The filters are usually washed with medium in a large Petri dish. Embryos are selected at a magnification of 12x - 50x using a simple stereoscopic viewer or a light microscope and transferred to dishes containing culture medium. As a substitute for the culture medium the medium is usually supplemented with 20 percent foetal or newborn calf serum. The addition of bovine serum albumin at a concentration of 3 mg/ml has proven effective especially when embryos are to be cryoconserved.
The embryos usually possess a diameter of approximately 0.16 mm. They are still surrounded by the zona pellucida, and have reached either the morula or blastocyst stage at the day of collection (day 6–8). Before the embryos are stored in an embryo bank it is of utmost importance to grade them qualitatively to guarantee good results after revitalization and transfer into suitable host animals. As summarized in Figure 6 grading of the embryos usually takes into account a description of the state of development (is it in agreement with the expected physiological state?), and the morphology (compactness of the cell mass). Optimal results are obtained if embryos are used that have been graded as “good” or “excellent”.
Embryos may be graded according to the following guide-lines:
| Excellent: | These embryos correspond to what may be expected from their stage of development. They are translucent with a yellow tint. Blastomeres are uniform in size. They are solidly attached to each other and no partially or completely detached cells are observed. The zona pellucida has not become invaginated. It is round and devoid of any particulate matter. The embryo does not float in the medium. Further development is observable after several hours of culture. |
| Good: | Embryos almost correspond to what may be expected from their stage of development. Blastocysts are uniform in size with a maximum of one having become detached. The zona pellucida is round and devoid of any particulate matter. Further development is observable after several hours. |
| Fair: | Appearance deviates up to 24 hours from expected developmental stage. Blastomeres are tinted yellow with dark spots. The tissue is solid with several detached blastomeres. Slight variation in size. The zona pellucida is not completely spherical. No further development discernable after several hours of culture. |
| Poor: | Embryo does not correspond to what may be expected from its stage of development and is usually 2–3 days retarded. Dark coloration and opaque appearance. Blastomeres loosely connected and varying in sizes. Several blastomeres have become detached. The zona pellucida is deformed and covered with particulate matter. Further development not discernable within 24 hours of culture. |
| Degenerated: | Blastomeres display large differences in size. Loosely connected tissue with individual disintegrated cells. Dark or black coloration. The zona pellucida is severely deformed. |
| Dead: | Complete disintegration of blastomeres. Black coloration. |
During freezing of pure water, the solvent is converted into a biological inert body, i.e. ice crystals, thus necessitating addition of anti-freeze compounds to prevent embryos from being damaged upon freezing and thawing.
| a) “excellent” | 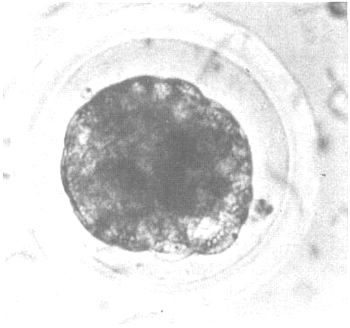 |
| b) “good” | 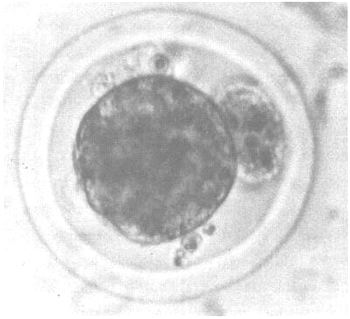 |
| c) “fair” | 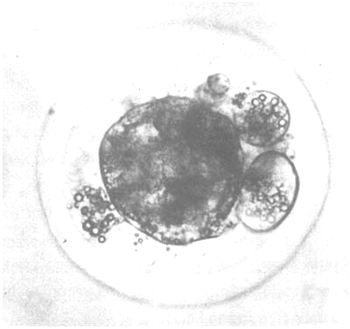 |
d) “poor” | 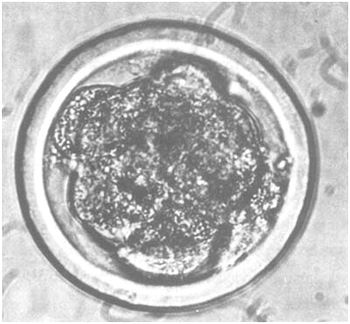 |
e) “degenerated” (unsatisfactory) | 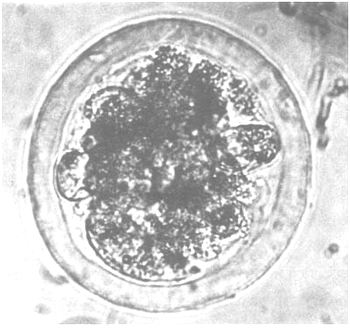 |
f) “dead” | 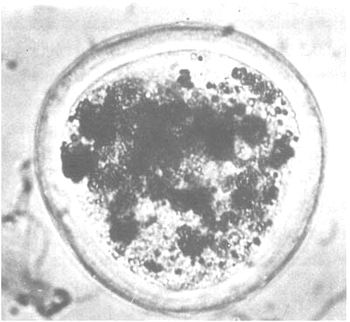 |
Figure 6: Quality of Cattle Embryos at Day 6
In principle there are two different groups of compounds that may be used for this purpose:
The cryoprotectant must be able to penetrate the cellular membranes to protect the cells. Examples of suitable compounds are glycerol, dimethylsulfoxide (DMSO), and low-molecular weight alcohols. Many of these substances are toxic to cells. Selection of the appropriate compound is therefore of utmost importance. Although DMSO has been established as a cryoprotectant for mouse embryos, glycerol appears to be best for cattle embryos, causing the least degree of damage. Effective working concentrations are usually in the order of 1–4 M.
Polyvinyl pyrrolidone, sucrose, glucose and other sugars are cryoprotectants that do not have to penetrate the cellular membrane. They are usually effective at low concentrations (0.01–0.2 M).
Cryoprotectants act in different ways. Firstly they minimize mechanical damage to cells brought about by crystal formation when the freezing point of the medium is lowered. Secondly they also strengthen cellular membranes in a way not yet understood. A plethora of techniques has been described for freezing and thawing cattle embryos. Most of them employ glycerol as cryoprotectant. Various concentrations have been used, but the most common concentration is 10 percent glycerol.
Glycerol is either added in several concentration steps, allowing the cells to equilibrate for at least 10 minutes, or in one step. Single embryos or groups are then stored in plastic straws and allowed to cool down from ambient temperature to -7°C. At this temperature crystallization of the freezing medium is initiated at some point distant from the cell masses (seeding). The embryos are again allowed to equilibrate for 5–10 min and are then cooled at a rate of 0.3–0.5°C per minute until the final temperature of -28°C to -35°C is reached. They are then transferred to liquid nitrogen. Provided that the individual specimens have been labeled appropriately the embryos may be stored in liquid nitrogen for decades. Neither the genome nor extrachromosomal genetic material will be altered in any way during this time.
The central problem with conserving embryos is the production of prime quality material from selected donor animals. Storage in liquid nitrogen has become a standard method, but further improvement of the freezing techniques can be expected.
Existing and well-planned embryo transfer programmes are a fundamental prerequisite for the development of techniques allowing manipulation of embryos. It was the establishment of embryo transfer techniques that allowed preimplantation manipulation of embryos in the first place. This also permitted the spectrum of available breeding techniques to be expanded considerably. Table 2 is a brief summary of embryonal stages currently used and of techniques employed for reactivating conserved genetic information.
| Embryonal stage | Technique | Chapter |
| Zygotes (fertilized oocyte) | Gene transfer by microinjection Nuclear transfer | 3.2.2 2.2.3 |
| 2–16 cell stage | Generation of chimaeras Monozygotic twins | 2.2.2 2.1.2 |
| Morulae & blastocysts | Generation of chimaeras by aggregation or injection of cells Monozygotic twins | 3.2.2.3 2.1.2 |
Table 2: Techniques for Micromanipulation of Preimplantation Embryos
Manipulation techniques for microsurgical separation of embryos and subsequent generation of identical twins can also be used for establishing embryo banks. According to the underlying concept two halves of split embryos rather than intact embryos should be frozen and stored separately. This approach allows two different strategies to be pursued:
Storage of both halves of a split embryo.
Both halves of a split embryo are frozen and stored immediately after having been
separated. From a genetical point of view this technique is identical to the
cryoconservation of intact embryos. A possible advantage for programmes designed at
reactivating the gene pool is the ability to generate more animals if further development
of individual halves proves to be successful.
Cryoconservation of one half of a split embryo and immediate transfer of the second half into a suitable recipient animal.
According to this strategy one half of the split embryo is transferred immediately into a suitable recipient animal. The other half is frozen and stored. If the transfer of one half results in pregnancy and the subsequent birth of a calf, the latter can be analysed and tested in detail. This approach provides a variety of data such as sex, body conformation, performance etc. for the genetically identical half that has been cryoconserved.
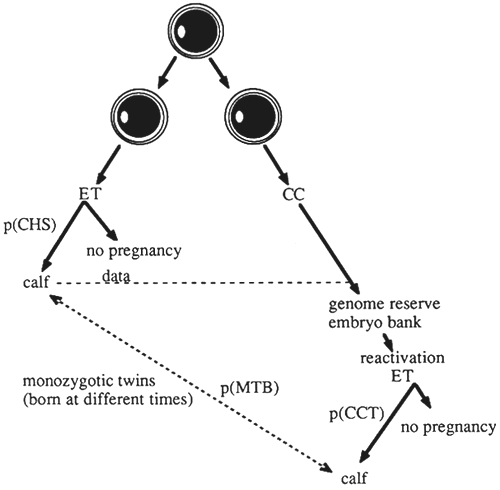
Figure 7: Cryoconservation of Split Embryos for the Production of Monozygotic Twins Born at Different Times.
ET = embryo transfer; CC = cryoconserved; p(CHS) = probability of obtaining a calf upon transfer of a demi-embryo; p(CCT) = probability of obtaining a calf upon transfer of a cryoconserved and thawed demiembryo; p(MTB) = probability of obtaining monozygotic twins born at different times.
This would be advantageous for the subsequent reactivation of the gene pool because the specimens to be thawed can be selected according to the data obtained. A mobile manipulation unit that we have been using for embryo splitting consists of a stereo microscope (Wild M8) with transmitted light illuminator and four micromanipulators (Figure 8).

Figure 8: Manipulation Set-up for Embryo Microsurgery
The microscope and the manipulators are fixed to a work-bench. The entire unit can be packed conveniently in two transport boxes and transported easily over long distances. It takes barely 30 minutes to set up the entire unit. The only important point is that a quiet and clean room, a stable bench, and electrical power should be available at the location where the embryos are to be split. The following microtools are required for the operation:
Holding pipette with drawn tip (outer diameter ca 110 μm; inner diameter ca 30 μm)
Microknife prepared from razor blades with sharpened tips
Glass needle with finely drawn tip (diameter ca 2 μm)
Finely drawn glass thread ending in a glass bead (diameter ca 30 μm)
Glass hook prepared from a finely drawn glass tip.
These microinstruments are fixed in suitable instrument holders. These can be fastened to the micromanipulators and positioned at the level of the Petri dish. The arrangement of the microinstruments in the viewing field is shown in Figure 9.
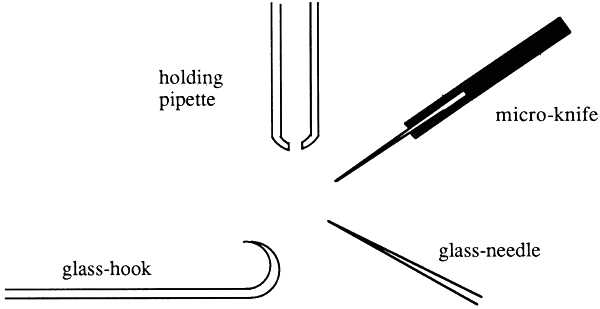
Figure 9: Schematical View of Microinstruments for Manipulation of Cattle Embryos
Microsurgical separation of cattle embryos is carried out preferably with compacted morulae or blastocysts (day 7). The embryo to be split is first positioned under the microscope in a Petri dish containing phosphate-buffered saline and 20 percent foetal calf serum. The holding pipette is used to fix the embryo by mild suction. Then the microknife is used to make a small incision in the zona pellucida of the embryo, and the fissure is subsequently propped open by means of the glass hook. This opening allows easy access to the embryo.
The embryo can be split either while being inside the zona pellucida or after having been removed from it. Splitting is achieved by a vertical cut using either the microknife, or the glass needle. After having separated the embryo into two halves, one half is re-inserted into the zona pellucida. The other half requires a suitable acceptor zona pellucida. It is usually prepared from an unfertilized oocyte or a degenerated embryo by opening its zona pellucida and removing the cell detritus. The second half of the split embryo are then inserted into the acceptor zona pellucida. The two halves of the split embryo are then removed from the manipulation dish and can be cryoconserved or used for transfer into an animal.
The nucleus of each cell of an organism contains the entire genetic information. This opens up the possibility - at least in theory - to conserve the genetic information of individual organisms in the form of cells or cell nuclei. Until now, however, it has been impossible to reactivate an entire organism from the genetic information contained in an individual cell. Exemptions from this rule are totipotent embryonal stem cells or blastomeres derived from very early embryonal stages. However, one may surmise that future developments in biotechnological research will rapidly advance these techniques. Since it can be assumed that the reactivation of genetic resources will be envisaged 15, 20, or more years from now, it seems justified to hope for developments in this field. In order to be able to exploit these possibilities in the future one should also store cells and/or cell nuclei as an alternative to storing genes.
Table 3 gives an overview of the suitability of cells or cell lines for conservation and reactivation.
| Cells | Work effort involved | Suitability of long-term storage | Reactivation potential |
| Early blastomeres | high | medium | good |
| ICM* cells | medium | good | good |
| Embryonal stem cells (EK cells) | very high | excellent | excellent |
| Teratocarcinoma cells (EC cells) | very high | good | good |
| Foetal cells (PGC)** | very high | excellent | excellent |
| Somatic cells | small | excellent | very low |
* ICM = inner cell mass of blastocysts
** PGC = primordial germ cells
Table 3: Suitability of Cells for Cryoconservation and Reactivation
• Blastomeres of early division stages
During the first four days following fertilization cattle embryos still reside in the oviduct. Their retrieval therefore requires the use of surgical techniques. Flushing of bovine oviducts entails great expense in terms of personnel, costs, and organization. Therefore this procedure cannot usually be carried out in the stables of the stock owner because a suitable room for surgery is required. An alternative would be the extraction of embryos after the donor animals have been slaughtered. However, especially as a means of establishing gene reserves for endangered cattle breeds, this will rarely have to be considered.
Long-term conservation can be achieved by storing either untreated embryos (See Section 2.1.1), blastocysts (following separation of the individual blastomeres), or karyoblasts (mini-cells containing the nucleus and being devoid of most of the cytoplasm). Early embryos may also be cryoconserved, although their survival rates are lower than those of later embryonal stages. No firm data are as yet available for isolated blastomeres and karyoblasts. However, the chances to reactivate individual animals from thawed blastomeres surviving this treatment are satisfactory.
• Cells from the inner cell mass of blastocysts
The extraction of blastocysts is a routine part of conventional embryo transfer programmes. The isolation of ICM (Inner Cell Mass) cells can be achieved by immunosurgical or microsurgical techniques. While the cryoconservation of untreated blastocysts is successful, the chances to conserve isolated cells are reduced, although the reactivation potential of surviving ICM cells is good or even excellent. According to current knowledge, storing individual cells will only have an advantage over storing entire embryos only in a very small number of special cases.
• Embryonal Stem Cells
Embryonal stem cells are pluripotent cells of a permanent cell line derived from an embryo. The process of establishing such cell lines is very time-consuming, and the establishment of such lines from cattle has not yet been reported. The technique deserves mention, however, because embryonal stem cells have been used widely in mouse systems. Strong efforts are being made with similar projects in cattle. The major advantage would be a very high reactivation potential of the genetic information contained in these permanent cell lines.
• Teratocarcinoma Cells
The properties of teratocarcinoma cells resemble those of embryonal stem cells. They are obtained from teratocarcinomas or from embryos which have been transplanted ectopically. As with embryonal stem cells no systems have as yet been established for cattle.
• Foetal Cells
Cell cultures may be established relatively easily from aborted foetuses or foetuses obtained after slaughtering. These cells can be conserved rather well. It must be pointed out, however, that according to currently available information the reactivation potential of these cells is relatively low.
• Somatic Cells
Somatic cells obtained from adult animals can also be used for establishing cell lines. Proliferation of the cells and the life span of the cultures in vitro, however, are limited. Cryoconservation in liquid nitrogen is relatively straightforward and yields satisfactory results. Yet, currently there is virtually no way to reactivate entire organisms from the genetic information contained in somatic cells.
It has been mentioned that according to current knowledge each of the approaches mentioned above still yields less satisfactory results than storage of entire embryos. New developments may, nevertheless, lead to improvements.
In order to prolong the life span and to produce larger quantities of semen ejaculates must be diluted before being used for insemination. The diluent must meet several important prerequisites.
It should
be isotonic with respect to semen plasma
buffer toxic products of metabolism
supply energy once thawed
possess anti-oxidative properties
be a good preserving agent
protect against shock associated with temperature shifts and dilution
inhibit bacterial growth
preserve semen in a biologically active form allowing fertilization.
Fresh semen can be stored at room temperature, in a refrigerator, or in a CO2 atmosphere for several days. Following programmed freezing, semen can be stored in dry ice (-79°C) or liquid nitrogen (-196°C) for decades. Cryoconservation of semen has been described for the first time by Polge, Smith and Parkes (1949) using chicken semen at low temperatures.
The difficulty of long-term conservation of semen in liquid nitrogen at -196°C is in minimizing damage by controlling the freezing process. Semen is usually damaged by the osmotic influence which is caused by water freezing first and thus progressively increasing the concentration of free ions in the immediate vicinity of the cells. This process is stopped with decreasing temperatures which also freezes residual liquids. Alterations in pH may rapidly denature lipoproteins. In addition it has been assumed that the plasma membrane of the semen cells loses lecithine. Depending on the quality of the semen preparation 5–20 percent of the cells in an ejaculate may not survive the freezing process. Mechanical damages produced by the formation of ice crystals may also occur, although it has less importance.
It has been assumed that the cryotic point of solidification may be the temperature boundary beyond which no further significant damage of semen cells is to be expected. Therefore the stress is greatest in diluted semen preparations which are in a partially liquid and partially crystalline state. It is therefore important to overcome this stage of the freezing process, which is reached at temperatures between 0°C and 12°C, as quickly as possible.
A variety of different crystals are produced at different low temperatures. They are produced consecutively with decreasing temperatures: first hexagonal, then cube-shaped, and finally amorphous forms. Fissures in the membrane of semen cells suddenly arising by recrystallization may lead to mechanical damage. It is therefore important to prevent recrystallization when semen preparations are stored for prolonged periods. Preferably, semen should be kept continuously either in liquid nitrogen or in nitrogen gas just above the level of the liquid at a temperature of approximately -130° C. Repacking of the contents of a container must proceed rapidly, and controlling individual vials of aliquot semen batches should be carried out either in liquid nitrogen or in nitrogen gas since a rise in temperature to -80°C to -75°C may already be critical.
Dilutions used for cryoconservation of semen depend on the different methods such as ampoules, straws, and pellets, and the doses used. A certain number of live semen per batch must be available after freezing and thawing to guarantee full fertilization capabilities. Under experimental conditions 3–5 million live semen cells will be sufficient, while 10–20 million live semen cells will yield excellent result in routine insemination programmes. Using more than 20 million live semen cells will not improve results significantly. In most breeding stations with a high frequency of insemination maximal usage of a bull's semen is guaranteed by diluting the semen to obtain doses with 12–15 million live cells.
The amount of viable cells present after cryoconservation is of paramount importance. Approximately 70 percent of the cells should be viable in ejaculates to be diluted. Good ejaculates usually contain 50–65 percent of viable cells after cryoconservation. Semen containing less than 50 percent of viable cells is not used for routine insemination and only in exceptional circumstances.
Ampoule Technique
Semen is treated as follows:
The semen is kept in a water bath at 25–30° C. Half of the total diluent required without glycerol (diluent a) is added to the semen. Amongst others Tris-or citrate deep-freeze diluent has proven particularly effective.
The diluted semen is cooled to +5°C within three hours by placing it in a refrigerator.
The second portion of the diluent (diluent b, containing glycerol) is added.
Glycerol concentrations are adjusted within six hours at +5°C. This period is usually referred to as equilibration time and may very in length.
The diluted semen is divided into ampoules, which are subsequently sealed.
Sealed ampoules are placed on a metal net approximately 4 cm above the level of liquid nitrogen for cryoconservation. Ampoules should be placed horizontally in one layer. Rotating the ampoules along the horizontal axis may accelerate the freezing process.
Recent investigations suggest that freezing with the help of specially designed programmable freezing equipment is unnecessary. The addition of glycerol to semen kept at +35°C or addition after cooling to +5°C is still a matter of discussion. However, the results obtained do not differ significantly.
Straw Technique
Sörensen (1940) was the first to use plastic vials for insemination purposes. Cassou (1952, 1968) put the technique into practice under the name of straw insemination. This technique was first used in France and has been adopted in Switzerland, Austria and Southern Germany. Its use is also common in Sweden, Norway and Japan. Straws are small plastic vials holding either 0.25 ml (mini-straws), 0.45 ml, or 1.2 ml. Straws holding 0.45 ml are used most frequently, but the mini-straw has also proven effective and will probably be used to a much wider extent because it takes up less space.
Straws have the additional advantage of guaranteeing that almost the entire dose of semen can be deposited in a cow provided a plastic catheter is used. In addition, straws are thin-walled and therefore stored semen usually shows better survival rates during the freezing process. This, in turn, allows for higher semen dilutions and hence a maximal usage of the bull semen.
Semen stored in straws is usually diluted so as to obtain approximately 15 million viable cells after thawing.
Freezing is frequently carried out in the presence of Laiciphos, a substance used to dilute milk, or in sodium citrate diluent. When Laiciphos is used the first half of the diluent usually contains 20 percent egg yolk. The second half of the diluent, which is added to the semen after cooling to 5°C, contains 14 percent glycerol instead. When mini-straws are used the diluent usually contains 10 percent egg yolk. Diluted semen is usually allowed to equilibrate for 4–7 hours (adaptation period) after the addition of glycerol. Other dilents such as Tris diluent may also be used.
Before use empty straws can be labelled individually, bundled, and exposed to UV light for sterilization. Straws are sealed after having been filled by dipping the open end into polyvinyl chloride alcohol. The process of filling and sealing by sonification has also been mechanized.
Straws are frozen without special precautions in large containers. They are positioned horizontally or vertically in a wire basket or on specially designed ramps. These are lowered into a container with liquid nitrogen so that the bottom touches the liquid. Straws will still be approximately 4 cm above the level of the liquid. Because nitrogen gas is also generated by partially dipping the basket into the liquid nitrogen, straws are usually frozen to -100°C within 5 minutes. After 7 minutes they may be immersed completely in liquid nitrogen.
If straws are required for insemination they are thawed by immersing them in water at 35– 40°C. The stopper is then cut away on one side and the straw is then positioned with the open side towards the tip of the insemination apparatus.
The technique described by Simmet (1972 a, b) employs plastic vials which are open at both ends. They are used in an automatic filling apparatus which, after having filled in the semen, inserts two small plastic balls at either side and seals them. Prior to insemination one of the plastic balls is removed by cutting off one end of the vial or by pressing the vial with two fingers. The other plastic ball serves as a piston for emptying the vial. The air bubble within the straw must be positioned in the middle of the vial to relieve pressure when the vial is frozen or thawed (danger of explosion!). The vials used are usually 90 mm in length and can hold 0.5 ml. Since the air bubble takes up some space, the effective volume available for semen is approximately 0.38 mL.
Pellet Technique
Semen is diluted at 30° C with a medium containing fructose, egg yolk, and glycerol. Tris or sodium citrate diluent have also been reported to yield satisfactory results. Due to the small size of the pellets (0.1–0.15 ml) semen is normally diluted only two- to four-fold, depending on the cell density of the ejaculate and the motility of the semen.
Diluted semen is precooled to +5°C and is then frozen after an adaptation time of 2–6 hours. Recent investigations have demonstrated that highest survival rates are obtained by a very short treatment with glycerol. Glycerol must be added together with a portion of the diluent and after temperature adaptation to +5°C immediately before pelleting. Pellets must be produced within ten seconds.
Freezing is achieved by using a CO2 ice disk in which small holes have been punched by a metal matrix which serve as moulds for the pellets. After approximately five minutes pelleted semen preparations can be removed and stored in liquid nitrogen. Individual pellets can be identified by small labels put on top of the semen before it is frozen. Contamination with microorganisms and easier identification is achieved by pelleting semen in small gelatine capsules (Capsule pellets).
For insemination pellets are put into thawing solutions prewarmed to 45°C where they will become liquid within seconds. Suitable solutions are thawing milk. Hannover's thawing solution, or sodium citrate solution.
Highest fertilization rates are obtained if pellets are used for insemination immediately after thawing.
Combi-pellets: Pellets of frozen semen are put on top of a frozen thawing solution in a plastic vial. The vial can also be sealed with aluminium foil and can then be despatched. Immediately before insemination the vial is heated to 45°C in a water bath to thaw the pellet and the diluent.
The pellet technique is easy to handle and does not require expensive equipment while yielding very good thawing rates. Freezing the pellets can be simplified by pelleting and testing out a portion of the ejaculate to be pelleted. Long-term storage is associated with a minimum amount of costs for packaging. Containers used for storing the pellets derived from and ejaculate may be reused.
Storage of semen preparations at a temperature of -196°C more or less arrests cellular metabolism. Several investigations have demonstrated that the ability of semen stored for several years in liquid nitrogen to fertilize oocytes is not reduced if at least 50 percent of the spermatozoa are motile at the time of insemination. This means that the individual doses prepared from an ejaculate can actually be stored for several years. However, the necessary motility required for insemination must be tested in the laboratory with one dose at least before use. Experience has shown that the mobility of high-quality semen is decreased only slightly upon prolonged storage. Loss of quality may be considerable in semen preparations which have been graded as satisfactory at the time of freezing.
The recovery of mature bovine oocytes requires surgery and is a rather laborious process. Oocytes from superovulated donors may be recovered either by flushing the oviduct after ovulation, or by aspirating mature Graafian follicles before ovulation. Suitable techniques have been worked out with the establishment of in vitro fertilization (IVS) programmes.
Since the recovery of ovulated oocytes for the purpose of gamete conservation during the establishment of gene reserves is not envisaged routinely, this topic will not be discussed in detail.
An alternative to the recovery of mature oocytes has been suggested by experiments designed to recover immature oocytes through aspiration biopsy of large follicles (∅ 1–5 mm) and subsequent maturation in vitro. Suitable ovaries are usually obtained from slaughtered animals. The technique must therefore be considered as a complement to other methods of conservation. Recovery of immature oocytes may be a way to conserve the genome of animals which cannot be used for the production of embryos, or which are otherwise unsuitable.
Collection of ovaries from slaughtered animals is unproblematic. They can be transported easily in isotonic sodium chloride solution at temperatures between 25–30° C in a thermos flask. Oocytes are recovered by puncturing all follicles at the surface of the ovary whose diameter is between 2–5 mm and by aspirating the fluid by means of a simple syringe with a fitted small canula (∅ 0.9 mm). With the help of a stereo-microscope (10x) individual oocytes can be selected and transferred from the follicle fluid of triple-strength modified Tyrode solution for washing. Only those oocytes are used which, upon vital staining with 0.2 percent trypane blue solution, show a compact cumulus oophorus and live cumulus cells. Two thirds of all cattle will usually yield ovaries suitable for the isolation of oocytes by aspirating follicles. A mean number of 25 follicles can usually be obtained from one animal, and approximately 5 intact live oocytes per animal are normally fit for further in vitro maturation.
A variety of media has been used for maturation of oocytes. One suitable technique is incubation in modified Tyrode solution (10 percent FCS, 11 μg pyruvate, and 10 mg FSH per μL of medium) at 39°C, 5 percent CO2, and a humidity of 100 percent for 24 hours. Even better results are obtained if oocytes are cocultivated with granulosa cells (106/ml). This technique allows more than 80 percent of the oocytes to complete meiosis (Lutterbach, Koll and Brem, 1987).
An alternative to the storage of mature oocytes is in vitro fertilization of oocytes and storage of zygotes (fertilized oocytes). Frozen semen can be used for in vitro fertilization. Fertilization results are better if spermatozoa are prepared by the so-called sperm swim-up technique (Parrish, Susko-Parrish and First, 1985). For this technique five oocytes and 2.5 × 106 spermatozoa are suspended in 50 μL of TALP medium (Tyrode's medium, albumin, lactate, pyruvate) supplemented with 5 percent FCS, 11 μg/ml pyruvate, 10 μg /ml heparin, 1 μM epinephrine, and 10 μM hypotaurine. This suspension is incubated under the same conditions used for maturating oocytes. Approximately 40 percent of the oocytes usually become activated and more than 30 percent become fertilized, as demonstrated by the occurrence of two pronuclei (Lutterbach, Koll and Brem, 1987).
A number of experiments has demonstrated that mature oocytes can be frozen and stored successfully in liquid nitrogen. Michaelis, Rubin and Hahn (1984) have conserved oocytes from CD2F1/Han mice with and without cumulus cells in liquid nitrogen by employing a 2-step freezing technique. The frozen cells were thawed by directly placing the vials in a water bath of 20°C. In either case approximately two thirds of the cells were morphologically intact after thawing. Subsequent in vitro fertilizations were successful in about 60 percent, while controls yielded 72 percent successes. Upon transfer into the oviducts of synchronized recipients 47.5 percent of oocytes successfully fertilized in vitro developed into foetuses (controls 50 percent).
Ridha et al. (1986) have recovered oocytes by aspiration from ovary follicles of camels. Upon storage in liquid nitrogen for 20 days 80 percent of the thawed oocytes were still viable. However, only 20 percent of them could be matured successfully.
Kiehm et al. (1987) have collected rat oocytes with cumulus cells 2–4 hours after ovulation and have removed the cumulus cells by treatment with hyaluronidase and washing in PBS containing 0.1 percent BSA. These oocytes were subsequently stored in straws and frozen in liquid nitrogen with DMSO or ethylene glycol as cryoprotectant. Following thawing and washing the oocytes were used for in vitro fertilization and cultivation. Almost 70 percent of all oocytes could be fertilized successfully, and 25–69 percent of all oocytes fertilized successfully underwent further development in vitro.
A much more involved technique for cryoconservation of oocytes has been suggested by Daniel and Tuneja (1987). They took ovaries from slaughtered cattle and cut them in longitudinal sections of 1 mm thickness. These were washed first in PBS containing 10 percent FCS and 15 percent glycerol and then placed in cryotubes containing a cryoprotectant. The sections were then cooled to -7°C, frozen further after the point of seeding had been reached, and finally stored in liquid nitrogen. Upon rapid thawing in a water bath (37°C) the slices were washed with PBS. In tissue culture these ovary slices gave rise to cells developing into monolayers. This suggests that oocytes may also survive this treatment.
In summary it may be surmized that the conservation of ovaries at low temperatures following recovery and maturation of oocytes from ovaries obtained from slaughtered animals is an appropriate technique. This approach opens up the possibility to resort to these materials in the future, when techniques will have been developed for reactivating the genetic potential contained within this material. It may be envisaged that thawed ovary slices might be transferable into suitable recipients to obtain oocytes which can be fertilized. Appropriate experiments have been carried out successfully with mice, although it must be stated that in these experiments pieces, halves and entire ovaries were transferred from donor animals to ovarectomized recipients (Baunack, Gärtner and Werner, 1988). The acceptor mice yielded normal litters representing the genetic material of the transferred ovaries.
Embryos are thawed either rapidly or slowly according to the course taken during freezing. Rapid or slow thawing is carried out by transferring the straws containing the embryos from the storage container to a water bath held at 30° C and 20°C, respectively. When the ice crystals have disappeared after approximately 30 seconds the cryoprotectant must be removed from the medium containing the embryos. This can be achieved in two different ways: according to one technique glycerol is removed by stepwise dilution, which is a rather time-consuming procedure. Another way is to prevent osmotic shock by adding sucrose during the dilution process by placing the embryos in a sucrose solution of low molarity (usually 0.5 M). Sucrose is then removed by transferring the embryos to culture medium after 10 minutes of equilibration. Embryos are graded morphologically before transfer (See Section 2.1.1). The use of plastic straws has allowed carrying out all steps in a so-called one-step-in-straw procedure.
As shown in Figure 10 the drop of freezing medium (usually 10 percent glycerol) containing the embryos is sandwiched between the thawing solution (usually 0.5 M sucrose). The embryos are frozen as described in Section 2.1.1. After thawing the straw is shaken briefly and inserted into the transfer cannula after equilibration for 10 minutes. The embryo is then transferred directly. This technique has been adopted widely because the frozen embryos are easy to handle and a high proportion of animals become pregnant.
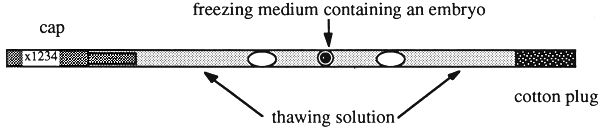
Figure 10: Preparing Straws for the One-Step-In-Straw Procedure
The transfer of embryos requires recipients whose oestrous cycle has been synchronized. At the day of embryo transfer these animals must be in the same stage of the oestrous cycle as the donor animals at the time of embryo collection. There are two different ways to achieve oestrous synchronization. One way is to transfer embryos six or seven days after the normal on set of oestrus. Another way is to synchronize the oestrous cycle. As shown in Figure 11 there are three techniques which have gained wide acceptance. A sound principle is to synchronize more recipients than required because individual animals may drop out for various reasons. The embryos are placed in a transfer cannula which resembles a pipette used for insemination and are transferred to the horn of the uterus which is positioned ipsilaterally to the corpus luteum. All manipulations of the uterus must be carried out with the utmost care.
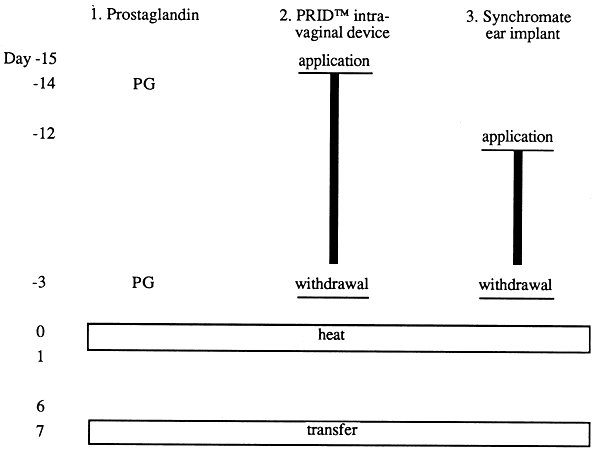
Figure 11: Oestrous Cycle Synchronization of Embryo Recipients
Several points have to be taken into consideration when choosing suitable recipients:
Heifers are more suitable than cows.
Recipients must be genitally healthy in every aspect.
Recipients must be of a breed that guarantees unproblematic births.
All veterinary and sanitary requirements must be fulfilled.
To a large extent the success rate of a programme depends on the quality of the recipients and the practical experience of the veterinarian carrying out the transfers.
Surgical transfer techniques employed initially have been replaced almost entirely by non-surgical methods. It has been demonstrated that the use of modern ultrasonic techniques is well suited to assess the state of the corpus luteum which had been thought possible only with the help of surgery.
Chimaeras are individuals which contain one or more cell types derived from two or more zygotes, germ cells, or cell lines. Individuals consisting of more than one cell type derived from a singular zygote are known as mosaics. Chimaeras are produced in the course of natural processes such as dispermy, aggregation of embryos, generation of vascular anastomosis, transplacental exchange of maternal and foetal cells, or by transfusion or transplantation.
Artificial mammalian chimaeras were generated only at the beginning of the 1960s (Tarkowski, 1961; Mintz 1962 a, b, 1964 a, b). Only few reports have been published describing the successful production of chimaeras in ruminants.
There are two fundamental techniques for generating chimaeras, i.e. the aggregation of embryos or parts of embryos and the injection of cells into the blastocoele, or into the inner cell mass.
Aggregation of embryos is a process by which whole embryos or parts are joined. The aggregation of genetically different embryos resulting in the formation of one embryo is one technique to obtain chimaeras. In principle the blastomere surface of different embryos is brought into contact at one or more points. The zona pellucida of each embryo is opened by microsurgery, in order to be able to manipulate the embryo. This procedure is similar to the technique employed for the production of monozygous twins. After separation and aggregation the embryos are again surrounded by the zona pellucida (Kräuβlich and Brem, 1985).
Separation and aggregation have to be carried out very carefully to prevent any damage to the cells. During further development the mixed populations of cells obtained by aggregation of cells with different genetic lineage will give rise to cell lines (clones). These, in turn, will develop into tissues with neighbouring cells possessing different genotypes.
The earliest embryonal stages which can be obtained from cattle by non-surgical techniques, i.e. by flushing of the uterus, are five days old. These embryos have reached the morula stage and contain 16 to 32 blastomeres. Separation of individual blastomeres by pipetting is not recommended at this stage. The mechanical stress exerted upon the cells would be too high because of the increasing strength of intracellular connections between individual blastomeres. These embryos are therefore split by means of a glass needle if halves rather than entire embryos can be used for the aggregation process. Sectioning of 5-day old embryos is usually associated with the loss of several blastomeres at the site of the section. The aggregation of embryos or its halves in a prepared empty zona pellucida is carried out in a way similar to that used for packaging embryo halves for the production of twins (Brem et al., 1983).
Figure 12 shows a cattle chimaera obtained by aggregating two morulae. The different contributions of Brown Swiss and Holstein-Friesian cattle are easily distinguished.

Figure 12: Chimaera between Brown Swiss and Holstein-Friesian Cattle.
The morulae used for aggregation were obtained by flushing the uterus at day 5 and were split in halves before use
Aggregation of parts of embryos and individual cells has been carried out successfully at least in experimental animals. It generally involves packaging of individual cells between two halves of an embryo. This increases the chances of the cells participating in further development of the embryo because the cells that were introduced are less likely to be excluded from the composite tissue produced by micromanipulation.
This technique has also been used with mice to generate chimaeras from embryos at the 8-cell stage and totipotent embryonal stem cells (EC-cells). Some of the resulting animals were also germ line chimaeras.
As shown in Figure 13, the injection technique used for the generation of chimaeras makes use of embryos at later stages of development, usually late blastocysts. The manipulation unit required for this procedure consists of a conventional set-up (microscope and two Leitz micromanipulators) in conjunction with a fine-adjustable injection system. This consists of an air-tight Hamilton microsyringe which is connected to the injection cannula by a thick-walled PVC rubber hose. The injection cannula is fastened to the manipulator. The entire system must be filled with paraffin oil free from bubbles to allow the precise controlled flow of medium at the tip of the injection cannula. The tip of the injection pipette must be cut at an angle and the cutting face must be sharpened. The walls should be as thin as possible with an inner diameter just large enough to give room for the injected cells.
The blastocysts and cells to be manipulated are placed in a manipulation chamber filled with medium and covered with paraffin oil. The blastocysts is then fixed to the holding pipette by slight suction. Several cells from the medium are taken up by the injection pipette at a short distance behind the tip. The pipette is then lowered into the blastocoele or the inner cell mass of the blastocyst by means of the micromanipulator. The cells are injected, and upon retrieval of the injection pipette care is taken that the cells do not escape from the blastocyst. The injected blastocysts can be cultivated in vitro and can then be transferred into suitable receipients.
Microinjection into the blastocoele has also been attempted to generate cattle chimaeras (Summers, Shelton and Bell, 1983). The haemogramme of one calf obtained in this way showed signs of participation of injected blastomeres. Another attempt to use microinjection of cattle blastomeres containing the marker chromosome 1/29 did not result in chimaeric animals (Stranzinger, 1983).
Cell populations in chimaeras are evenly distributed between the two extremes rather than showing a binomial pattern of distribution. In contrast, mosaics show a binomial distribution of market cells. The reason for the much larger variability in chimaeras is that only a single event, i.e. X-chromosome inactivation, will influence the distribution of both component cells in each individual in mosaics. In chimaeras there will be two events, namely differentiation into inner cell mass and trophoblast, and differentiation into primary ectoderm and endoderm.
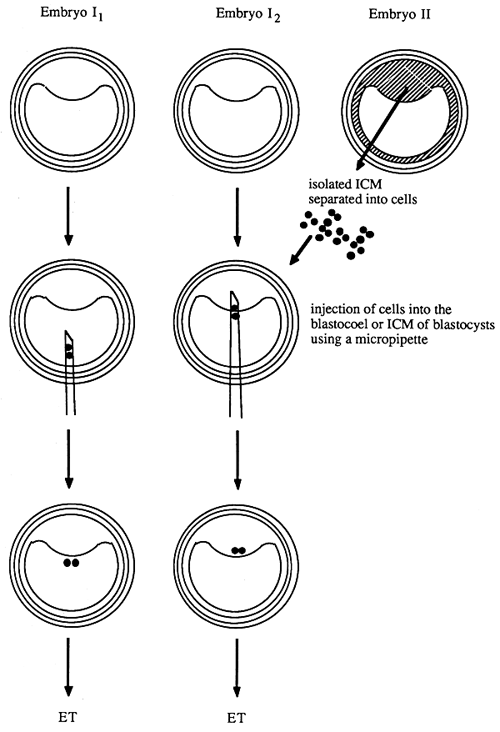
Figure 13: Generation of Chimaeras by Injection of Cells (from Embryo II) into the Blastocoel (Embryo I1) or the Inner Cell Mass (Embryo I2) of Blastocysts
The use of totipotent cells is of critical importance if chimaeras are produced by reactivating conserved cells. Apart from creating chimaeric somatic tissue, it is even more important to guarantee that gonads either entirely or at least in part will consist of thawed cells. Only then will the conserved genome be able to become reactivated in the next generation.
If the creation of chimaeras proves impossible it will be necessary to attempt the reactivation of the genome of conserved cells or blastomeres by nuclear transfer. In the course of experiments designed to clone animals, transplantation of nuclei derived from amphibian cells has been attempted for the past 35 years. Briggs and King (1952) were the first to transfer nuclei from blastula cells into enucleated frog oocytes, which later developed into normal embryos. Subsequently some successes have also been attained with transplantation of nuclei derived from somatic cells of the intestinal tract of tadpoles as well as skin, lymphocyte and blood cells of adult frogs. While the transfer of nuclei derived from the epithelial lining of the intestine of tadpoles has given rise to fully developed frogs, all other attempts have yielded embryos which died at the larval stage.
Figure 14 shows the principle of nuclear transfer as applied to mammals. It can be seen that the donor of the nucleus is usually an embryo. Nuclei from blastomeres derived from early embryonal stages are almost totipotent. In principle it should also be possible in future to use other nuclei. The problem with nuclei derived from more differentiated cells is that the DNA in the genome of these cells may undergo alterations with progressive differentiation, which may limit the totipotency of these nuclei. It is to be hoped that these alterations in nuclear DNA may be reversible, at least in part, by the development of new techniques.
Acceptors for the nuclei to be transferred are enucleated oocytes which must be prepared accordingly. Enucleation of fertilized oocytes can be achieved by microsurgery. By treating the oocytes with cytochalasin B the cytoskeleton of the cell can be altered in a way that results in the formation of very soft cellular membranes. A specially prepared micropipette can be inserted to reach the pronuclei of the cell without damaging the membrane of the pretreated oocytes. The pronuclei and the surrounding cellular membrane can be taken up into the lumen of the micropipette by careful suction. Retracting the pipette will remove the pronuclei from the cytoplasm of the cell. The cytoplasmic bridge which is created during the retraction of the pipette is broken when the pipette passes through the zona pellucida, generating an enucleated oocyte with an intact cell membrane.
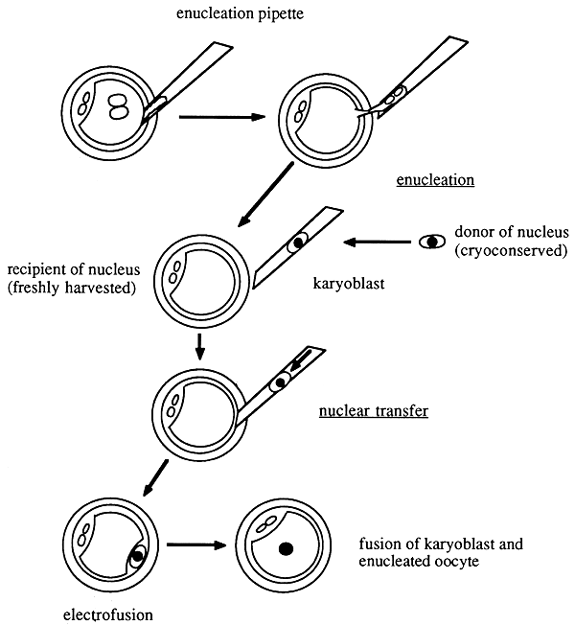
Figure 14: Enucleation and Nuclear Transfer
The most critical part of the nuclear transfer experiment is the last step, i.e. the transfer of the nucleus into the cytoplasm of an enucleated oocyte. Initial experiments have usually involved careful penetration of the pipette which had been used for enucleation to transfer the nucleus through the cellular membrane of the acceptor cell. It has been shown that this manipulation is rather delicate and that the majority of the oocytes do not survive the penetration process. Similar experiences have been made when oocytes were encleated by direct insertion of the enucleation pipette into the cytoplasm.
Illmensee and Hoppe (1981) were the first to transfer mamalian nuclei successfully. They used a technique which allowed enucleation of pronuclei and transfer of donor nuclei in a single step by first inserting a transfer pipette already containing a nucleus into the cytoplasm, injecting this nucleus and removing the pronuclei from the cytoplasm with the same pipette. By reducing the entire manipulation to a single penetration process they achieved good survival rates of the manipulated oocytes. Illmensee and Hoppe (1981) have reported the birth of three normal mice, generated by the transfer of nuclei derived from blastocysts. It should be noted, however, that they have not been able to reproduce their results. It may be that the technique is too complicated and too little advanced to be reproducible.
A new technique for nuclear transfer has been developed in the meantime. It is easier to handle and more successful with a variety of cell types and nuclei.
McGrath and Solter (1983) have used micromanipulation techniques to inject karyoblasts through the zona pellucida into the perivitteline space. Karyoblasts are nuclei with an intact nuclear membrane, which are also surrounded by a small fraction of cytoplasm. These karyoblasts were obtained from fertilized oocytes and were fused with enucleated cells by means of inactivated Sendai viruses. This technical improvement prevents damage of the cellular membrane during nuclear transfer. The survival rates of embryos have been higher than 90 percent and more than 90 percent of the manipulated embryos have developed into morula and blastocyst stages in vitro. Upon transfer of these embryos 16 percent have developed into normal mice.
In contrast to the technique described above which employs nuclei derived from zygotes, corresponding experiments with nuclei of multi-celled embryos and blastocysts have yielded unsatisfactory results in the mouse system (McGrath and Solter, 1984). An adoption of the karyoblast technique to farm animals may hold some promise.
The fusion of karyoblasts and enucleated oocytes by Sendai virus has gradually been replaced by electrofusion. A fusion apparatus developed by Zimmermann allows short electric pulses (20–40 ms, 100 V) to be used for fusion of cellular membranes. Robl et al. (1987) have transferred pronuclei of cattle oocytes into enucleated oocytes and fused them into the cytoplasm by electrofusion. The transfer of pronuclei yielded 17 percent morula and blastocyst stages. Embryos with nuclei derived from cells in the 2-cell and 8-cell stage did not develop further during in vivo culture employing sheep oviducts. Barnes et al. (1987) have enucleated mature oocytes and transferred nuclei of 4-cell to 16-cell stage embryos by dielectrophoresis; 27 percent of the cloned embryos developed in vivo into morula and blastocyst stages.
Willadsen (1986) has investigated the ability for further development of blastomeres obtained from 8-cell to 16-cell embryos by transferring these blastomeres into oocyte halves which has or had not been enucleated by using either Sendai virus-mediated fusion or electrofusion. More than 40 percent of the nuclei transferred into enucleated oocytes developed into blastocysts if the electrofusion technique was used. Several of these transferred blastocysts developed into healthy lambs, including a pair of monozygous twins. Some blastomeres derived from the 16-cell stage also possessed the capacity for full development. Press reports seem to suggest that Willadsen has also succeeded in using this technique with cattle (Cooke, 1986).
The experimental results described above demonstrate that the technical side of nuclear transfer has been developed to a stage which allows reproducible and successful nuclear transplantation. There are no important indications which suggest that the transfer of cryoconserved and thawed nuclei or karyoblasts might be associated with severe problems (See also Section 2.1).
It is much more difficult to decide upon the chances of nuclei derived from later embryonal stages to develop into fully grown healthy individuals. It may only be said safely that early embryonal stages in cattle have a good capacity for further development.
In the near future there is no chance of exploiting the genetic potential of somatic cells for reactivation. According to current knowledge it seems very unlikely that it will be possible to obtain entire organisms by reactivating either fresh or conserved differentiated somatic cells.
![]()
![]()
![]()