![]()
![]()
![]()
The first step in a detailed molecular analysis of the entire genome of an organism and hence the isolation of certain coding regions of a single chromosome is the isolation and enrichment of individual chromosomes, which has recently become feasible.
While commonly practised techniques of chromosome analysis preclude the isolation of genetic material (DNA), application of flow-through cytophotometry allows both karyotype analysis and recovery of sufficient amounts of material by chromosome sorting.
Flow-through cytophotometry essentially involves measurement of fluorescence emitted from chromosomes stained with suitable fluorescent dyes after excitement with monochromatic laser light. Fluorescent emissions are detected with light detectors and processed as electronic signals by a computer (Figure 15).
Apart from providing flow-through cytogrammes, the technique also allows determination of the amount of DNA per chromosome, and of base composition, and the degree of chromatin condensation. The suspension of chromosomes passes the laser beam in tiny droplets. These may be charged electrically according to the amount of fluorescence measured which, in turn, corresponds to individual chromosomes, or to a certain chromosome subset. The charged droplets can be deflected from their normal paths in an electric field, while the paths of uncharged droplets will not be affected. Enrichment of certain chromosome subsets by flow-through cytophotometry is also known as fluorescence-activated cell sorting.
A sorting frequency of approximately 100 specific chromosomes per second will yield ca 400000 enriched chromosomes per hour. In some cases the purity of the enriched chromosome fractions may reach 99 percent (Lebo et al., 1984). Normal values are usually in the order of 80 percent (Cremer and Cremer, 1985). Sorted chromosomes can be stored in suspension and may also be processed immediately.
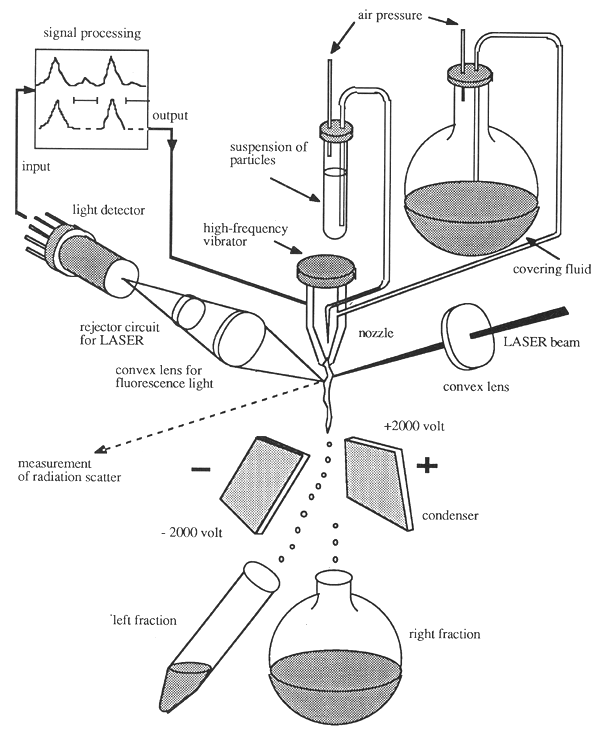
Figure 15: Schematic Representation of Fluorescence-Activated Cell and Chromosome Sorting (FACS) (Cremer and Cremer, 1985)
Other techniques, e.g. in situ hybridization to metaphase chromosomes, have allowed certain genes and gene clusters to be mapped to individual chromosomes. Chromosome sorting techniques have also facilitated the establishment of chromosome-specific DNA libraries.
In order to reach a high precision in chromosome sorting and to allow the better identification of individual chromosomes, a number of additional physical characteristics have been adopted as sorting parameters (Gray et al., 1979; Lebo et al., 1984). The yield of sorted chromosomes may also be increased considerably by employing methods allowing preenrichment such as sedimentation and other centrifugation techniques (Collard et al., 1984)
Another way of establishing chromosome-specific DNA libraries is provided by inter-species hybrids. This technique is based essentially on the fusion of cells stemming from two different species and a loss of chromosomes derived either from one or other species after cultivation of the cell hybrids under suitable conditions. Eventually viable and culturable cells are obtained which contain one or few chromosomes of one species as additional chromosomes. These cell hybrids can be used for establishing genomic libraries containing chromosome-specific recombinant clones.
Both techniques described above possess characteristic advantages and disadvantages with respect to fidelity. This means that a combination of chromosome fractionation techniques and cell hybridization methods must be used to obtain high-grade chromosome-specific DNA libraries.
Numerous examples have demonstrated that isolated chromosomes may also be used directly for gene transfer. The technique is known as chromosome-mediated gene transfer (CMGT) (Klobutcher and Ruddle, 1979; Willecke and Ruddle, 1975; Athwal and McBride, 1977; McBride and Ozer, 1973; Miller and Ruddle, 1978).
Gene libraries are collections of recombinant DNA molecules consisting of genomic DNA fragments of an organism and vector DNA. These molecules can be introduced into bacteria, where they can be multiplied and stored indefinitely. At best one obtains a genomic DNA library consisting of a number of individual clones which may easily be handled and which contain all possible DNA sequences of the genome in a stable manner in the form of overlapping DNA sequences.
Cloned DNA fragments should be long enough to contain complete genes together with their flanking sequences. On the other hand they should be small enough to allow restriction site mapping. In addition, it should be possible to establish a gene library from minute amounts of starting material, and to screen such a library for a desired sequence. Screening usually involves hybridization with radioactive DNA or RNA probes.
The size of a genomic DNA library which guarantees that a single sequence will be contained in the library depends upon the size of the cloned DNA fragments and the size of the genome. The number of independent clones, N, which must be screened to find a sequence with the probability, P, is
| N = ln(1 - P)/ln[1 -(I/G)] | (3) |
with I representing the size of the cloned DNA fragments (in base pairs), and G the size of the entire genome (in base pairs) (Clarke and Carbon, 1976).
In order to find a desired sequence with a probability of 99 percent the number of recombinant clones in a mammalian library established in a typical λ vector will be:
| N = ln(1 - 0.99)/ln[1-(2 × 104/3 × 109)] = 690000. | (4) |
The quality of a library can be evaluated by taking into account the average lengths of inserted DNA fragments, (I), and the number of clones obtained (N). The product, (I×N), should be 4.6 times the number of base pairs of the total genome (G) (Seed, Parker and Davidson, 1982).
The equation given above requires the cloned DNA fragments to be a random mixture of DNA sequences obtained from the genome. This prerequisite is fulfilled if the genome has been fragmented randomly for the construction of the library.
In a strict sense randomness can only be guaranteed if the genome has been “sheared” mechanically, e.g. by sonification. The techniques available for cloning “sheared” DNA are relatively inefficient and therefore use is made of a limited digestion with restriction enzymes to produce a mixture of DNA fragments, because this also produces a suitable population of random DNA fragments. For this purpose one usually employs limited digestion with restriction enzymes which recognize cleavage sites consisting of four nucleotides, i.e., enzymes which cut frequently. In principle it is also possible to digest partially the DNA with restriction enzymes with hexanucleotide recognition sequences. If the DNA in question is assumed to consist of 50 percent A+T and 50 percent G+C, a random hexanucleotide would occur every 4096 base pairs. This would guarantee a sufficient amount of randomness in the resulting DNA fragments. However, since a DNA composition of 50 percent A+T and 50 percent G+C is rather rare, use of these enzymes would lead to certain restriction fragments being under-represented and others being over-represented (Collins and Brüning, 1978).
Suitable enzymes recognizing tetranucleotide sequences are Sau3AI (/GATC), MboI (/GATC), AluI (AG/CT), and HaeIII (GG/CC). One may expect cleavage sites for these enzymes to occur every 256 base pairs. If these cleavage sites were distributed unevenly, one would still obtain a collection of DNA fragments upon hydrolysis of the DNA in which individual fragments of the desired length would be represented equally.
Vectors for Genomic DNA Libraries
Genomic DNA libraries are usually prepared in λ or cosmid vectors because they allow cloning of large size DNA inserts and because they possess high cloning efficiency. Plasmids such as pBR or pUC derivatives are not suitable because they allow efficient insertion of relatively small DNA fragments only and subsequent introduction and propagation of the vectors in bacteria is insufficient.
Derivatives of bacteriophage λ have been widely used to establish gene libraries. These derivatives have been altered by mutation, substitution, or deletion of certain regions of the λ genome to produce cloning vectors with restriction sites suitably placed for cloning purposes. A number of different λ cloning vectors are currently available (Table 4). They differ in their cloning capacities and in the presence of individual selective markers, and restriction cloning sites. The normal size of inserts that can be cloned without problems in λ vectors lies between 18–25 kb (Williams and Blattner, 1979; Tiemeyer, Enquist and Leder, 1976). One major advantage is the fact that recombinant DNA containing λ and genomic DNA sequences can be packaged in vitro to produce infectious phage particles which may be used for subsequent infections with high efficiency.
The reintroduction of naked λ DNA in vitro into empty phage heads has been described for the first time by Hohn (1979). Empty phage heads and other phage proteins engaged in the formation of mature phage heads can be obtained from two isogenic strains of E. coli known as BHB2688 and BHB2690.
These strains contain a temperature-sensitive lysogenic prophage which carries defects in two different genes required for morphogenesis. It is possible to isolate the necessary phage proteins and prehead structures required for the formation of mature phage heads from each strain. After in vitro packaging of recombinant phage/genomic DNA the mature phage particles can be used for infections. One advantage of λ vectors is the fact that storage and screening of a library are easy and straightforward.
| Vector | Size (kb) | Cleavage sites | Cloning capacity (kb) | Recombinant phenotype | References |
| Charon | 45.3 | EcoRI | 7.1–20.1 | lac-, bio- | 1) |
| 4A | XbaI | 0–5.63 | red-, gam- | 1) | |
| λgt WES | 40.4 | EcoRI | 2.2–15.1 | 2) | |
| λB | |||||
| λ1059 | 44 | BamHI | 6.3–24.4 | spi- | 3) |
| λgt11 | 43.7 | EcoRI | 0–4.8 | cIts, int+, red+ | 4) |
| λgt10 | 43.34 | EcoRI/HindIII | 0–5 | cI-, int-, red+ | 5) |
| λEMBL3 | 42.2 | BamHI/SalI/EcoRI | 10.4–20 | spi- | 6) |
| λ2001 | 41.2 | EcoRI/HindIII/SacI BamHI/XbaI/XhoI | 10.4–20 | spi- | 7) |
1) Blattner et al., 1977
2) Tiemeyer, Enquist and Leder, 1976
3) Karn et al., 1980
4) Young and Davis, 1985
5) Huynh, Young and Davis, 1985
6) Frischauf et al., 1983
7) Karn et al., 1984
Table 4: Characteristics of Commonly Used λ Vectors
A disadvantage is the fact that in vitro packaging is associated with a size selection of the DNA. λ DNA molecules must be packaged a new after each growth cycle. This means that phage recombinants whose size is near the lower or upper border of DNA that can still be packaged may in the long run be under-represented in libraries (Feiss et al., 1977). Another frequently used λ phage for the establishment of genomic libraries is EMBL3 (Frischauf et al., 1983).
The elucidation of the function of the so-called cos sites (abbr. of cohesive site) has been a major step in the investigation of the λ genome (Wu and Taylor, 1971). Cos sites are single-stranded DNA regions consisting of 12 complementary nucleotides which can be found at the 5' and 3' ends of the linear phage genome. They function as packaging signals which allow adjacent DNA to be packaged correctly into phage heads. This mechanism has been exploited for the construction of a new vector system which combines the advantages of plasmid and phage cloning. The resulting vectors which have been named cosmids contain an origin of DNA replication, one or two antibiotic resistance genes derived from a plasmid, and λ cos sites. The length of cosmids is usually in the order of 8 kb (Figure 16). Their small size allows DNA fragments with a length of up to 48 kb to be inserted into the vector, thus reducing considerably the number of clones required for a representative genomic library. Cosmids are the vectors of choice if large gene clusters such as MHC genes are to be investigated.
Cosmids of the first generation have been used to construct other cosmid vectors which, in addition to plasmid-derived replication origins, bacterial antibiotic resistance genes, and cos sites, also carry genes allowing transfection and selection in eukaryotic cells (Grosveld et al., 1982; Lau and Kan, 1983) (Table 5). These cosmids are also known as shuttle cosmids. Transfected genes can be reisolated from tissue-cultured cells (Lund, Grosveld and Flavell, 1982).
Wahl et al. (1987) have constructed cosmids (pWE2, pWE4, pWE8, pWE10, pWE15, pWE16; WE = walking easily) whose restriction cloning sites are flanked by phage promoters SP6, T7, and T3. These vectors allow in vitro synthesis of RNA probes if phage- and promoter-specific RNA polymerases are used. These RNAs may be used as probes for screening libraries or as starting material for chromosome walking. In addition the recognition sites of NotI (GC/GGCCGC) and SfiI (GGCCNNNN/NGGCC) have been cloned 5' or 3' to other cloning sites. These two cleavage sites are very rare in eukaryotic DNA and the use of these vectors therefore allows inserted DNA fragments to be recovered in an intact unfragmented form. The choice between phage and cosmid vector usually depends on the desired size of the genomic DNA inserts. Because handling of phage vectors is easier than handling cosmids the former are usually preferred, which means that fragment sizes of cloned DNA is usually in the order of 20 kb.
Partial hydrolysis of DNA during restriction digestion may be obtained by limiting the reaction time, or by limiting the amount of available enzyme. Irrespective of the technique used the DNA fragments produced early during reaction are more random than those occurring at later times (Seed, Parker and Davidson, 1982).
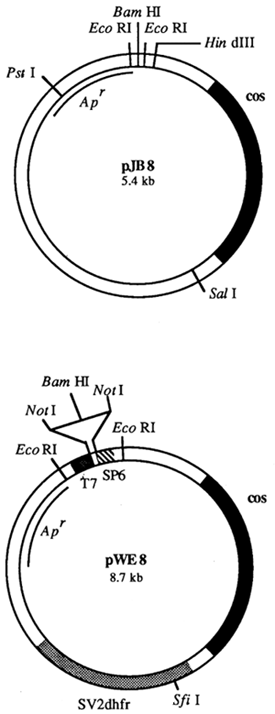
Figure 16: Structure of Cosmids pJB8 (Ish-Horowicz and Burke, 1981) and pWE8 (Wahl et al., 1987)
Apart from cos sites cosmid pJB8 carries the ampicillin resistance gene (Apr) and the origin of DNA
replication of pMB1. This plasmid can therefore be amplified by stimulation with chloramphenicol. The
BamHI site which is flanked by two EcoRI sites is usually employed for cloning foreign DNA.
Cosmid pWE8 is a so-called shuttle cosmid. It also contains two promoters derived from bacteriophages
T7 and SP6 thus allowing its use in in vitro transcription. The BamHI cloning site is flanked by two
NotI sites.
| Vector | Size (kb) | Cleavage sites | Antibiotic marker | Cloning capacity (kb) | Ori | Markers for use in eukaryotic cells | Ref. |
| 1. Prokaryotes | |||||||
| pJC 720 | 25 | HindIII | rif, ColE | ColE1 | 1) | ||
| pHC 79 | 6.4 | EcoRI/BamHI/ClaI/ HindIII/SalI | amp, tet | 32–44 | pMB1 | 2) | |
| pJB 8 | 5.4 | BamHI/HindIII/SalI | amp | 32–45 | pMB1 | 3) | |
| pcos2 EMBL | 6.1 | BamHI/XhoI/SalI | neo, tet | 31.7–44.8 | R6K | 4) | |
| MUA 3 | 4.76 | EcoRI/PstI/PvuII/ PvuI | amp, tet | 40–48 | pMB1 | 5) | |
| Homer I | 5.44 | EcoRI/ClaI | amp | 30–47 | pMB1 | 6) | |
| Homer II | 6.38 | SstI | amp | 32–44 | pMB1 | 6) | |
| Loric | 6.3 | BamHI/HindIII/ ClaI/XbaI | neo | 31.5–44.3 | λ | 7) | |
| 2. Eukaryotes (“shuttle cosmids”) | |||||||
| pOPF | 8.0 | BamHI/ClaI/KpnI | amp | 30–50 | SV40 | HSV-tk | 8) |
| pWE2 | 8.63 | BamHI | amp | 33–44 | SV2 | neo | 9) |
| pCV103 | BamHI | amp | SV40 | gpt | 10) | ||
| pPCV003 | 12.9 | EcoRI/BglII/SalI/ HindIII/BamHI | amp, cm, tet | RK2 | Kana | 10) | |
| pTM | 7.6 | BamHI/HindIII/ClaI | amp | G418 | 8) | ||
amp: ampicillin resistance gene
tet: tetracyclin resistance gene
cm: chloramphenicol resistance gene
neo: neomycin resistance gene
Kana: kanamycin resistance
gpt: guaninphosphoribosyl-transferase
HSV-tk: Herpes-Simplex-Virus thymidin-koinase
1) Collins and Brüning, 1978;
2) Hohn and Collins, 1980;
3) Ish-Horowicz and Burke, 1981;
4) Poustka et al., 1984;
5) Meyerowitz et al., 1980;
6) Chia, Scott and Rigby, 1982;
7) Little and Cross, 1985;
8) Grosveld et al., 1982;
9) Wahl et al., 1987;
10) Lau and Kan, 1983
Table 5: Characteristics of Frequently Used Cosmid Vectors
Restriction enzymes recognizing a tetrameric cleavage site produce a random mixture of DNA fragments which is more representative than fragments obtained by digestion with enzymes possessing hexameric cleavage sites. A general strategy is to select those enzymes which cut most frequently. In addition one must take care that the DNA fragments produced by enzymatic cleavage can be ligated into the vector that will be used for cloning.
Sau3A and MboI are the most frequently used enzymes. They recognize the sequence GATC and therefore cut the genome frequently. Fragments generated by these two enzymes can be ligated with BamHI-cut vectors and cosmids. The establishment of a genomic library in a bacteriophage vector usually requires 1 mg of high-molecular weight genomic DNA to be available for partial digestion with enzymes.
Control digestion can be used to find optimal reaction conditions. Partially digested DNA normally contains DNA fragments with sizes ranging between 100 bp and over 100 kb.
Depending on the vector used, partially digested DNA must be size-fractionated before ligation. Random mixtures of DNA fragments with a length of approximately 20 kb are prepared for use in λ vectors. If cosmid vectors are used, fragments of 40 kb are selected. Size selection of DNA mixtures can be carried out in linear sucrose gradients (5–40 percent) or NaCl gradients (1.25–5 M). In order to prevent unwanted recirculation or catenation of genomic DNA fragments cleaved DNA preparations are usually treated with alkaline phosphatase.
During ultracentrifugation the DNA fragments migrate according to size. The bottom of the centrifugation tube will be of the highest density and hence DNA with the highest molecular mass. Gradients are normally dripped by punching a small hole into the bottom of the tube. This also prevents cross-contamination of high- and low-molecular weight fragments.
Fractions containing suitably sized DNA fragments can be ligated with prepared vector DNA. The preparation of vector DNA is exemplified in Figure 17 with cosmid vector pJB8 (Ish-Horowicz and Burke, 1981).
The first step is to separate the cleavage of pJB8 (with HindIII or SalI) and subsequent treatment with alkaline phosphates, which prevents recirculation. The resulting DNA fragments are then cleaved with BamHI; the resulting long BamHI fragments are then mixed. Genomic DNA which has been cut with Sau3A or MboI can then be mixed and ligated with the vector DNA fragments; 0.5 mg partially digested genomic DNA may yield 5–10 μg of size-fractionated DNA.
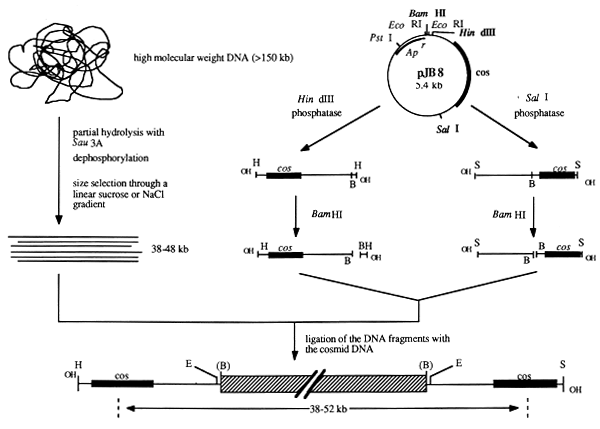
Figure 17: Establishment of a Cosmid Library in Vector pJB8 (Ish-Horowicz and Burke, 1981)
Vector pJB8 is linearized with HindIII and SalI and subsequently dephosphorylated with alkaline phosphatase in order to prevent recirculation. Further cleavage with BamHI generates vector arms which can be purified. These are then mixed and used for cloning genomic DNA which has been partially digested and size selected.
Another approach for the establishment of genomic DNA libraries is the use of Charon vectors which will be described below (Figure 18).
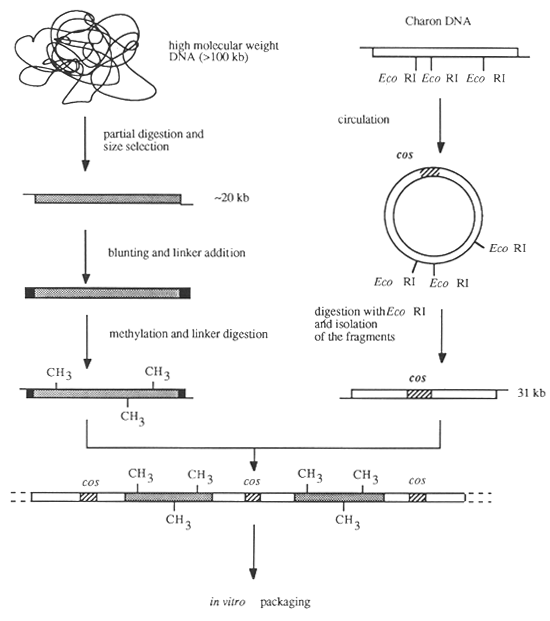
Figure 18: Establishment of a Genomic DNA Library in a Charon Vector
High-molecular weight DNA is prepared either from tissue or from blood. The use of material relatively devoid of connective tissue such as liver as starting material is an advantage. Suitable tissue samples can be obtained either as biopsy material from live animals or immediately after slaughtering. It is important that the material be processed immediately if no suitable means of conservation are available. Otherwise the tissue may also be stored at -20 °C, -80 °C or in liquid nitrogen for an indefinite period. Establishment of a genomic DNA library requires several milligrammes of tissue. If the DNA is to be isolated from blood, leucocytes should be prepared first. This can be achieved by centrifugation either in Ficoll-Paque gradients, or by simple centrifugation if large volumes (several litres) are to be processed.
Since high-molecular weight DNA (Mr = 3×107) is required for the preparation of genomic libraries, the DNA must be treated very carefully. Suitable techniques have been developed by Gross-Bellard, Oudet and Chambon (1973) and Blin and Stafford (1976). Both techniques will yield DNA with molecular masses between 4 × 106 and 500 × 106, which can be cleaved subsequently with restriction endonucleases.
Naked recombinant phage or cosmid DNA cannot be introduced satisfactorily into bacteria. It is therefore necessary to package the DNA into empty phage heads in vitro and to infect bacterial cells subsequently (Figure 19). This technique has been described for the first time by Hohn and Murray (1977) and Hohn (1979).
The principle of in vitro packaging is based essentially on the ability of DNA fragments carrying λ cos sites at their 5' and 3' ends to be packaged in vitro in the presence of empty phage heads and packaging proteins. This process requires that the molecules are at least 38 kb in length and not longer than 52 kb. Packaged phage heads are matured into infectious phage particles in vitro in the presence of gene products W and FII and phage tails. All proteins required for packaging can be obtained from strains BHB2688 and BHB2690 of E. coli as described above. Packaging mixes are prepared once and can be stored at -80 °C.
Depending upon the vector there are a number of E. coli strains which may be used for transfection (Table 6). These strains are starved before use, either by growth in minimal media and subsequent treatment with 0.1 M CaCl2 solution (Mandel and Higa, 1970), or by washing in 0.1 M CaCl2 solution after propagation in L-broth medium (Hohn and Murray, 1977). This pretreatment induces the synthesis of λ receptors (lamB protein) on the cell surface and significantly increases the absorption of the phage to E. coli cells.
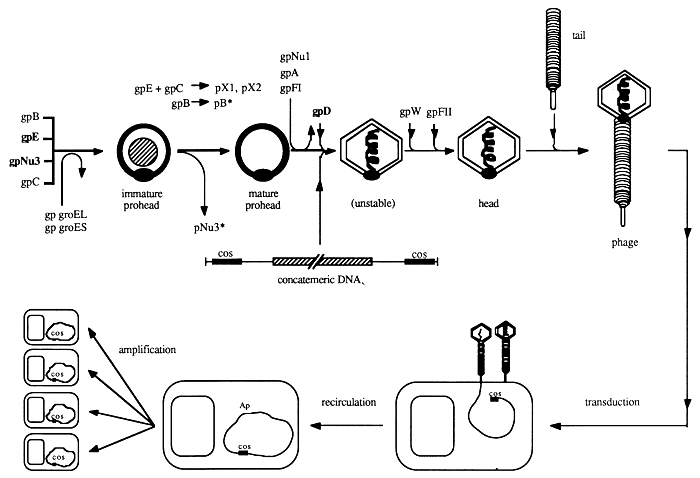
Figure 19: In vitro Packaging (Hohn, 1979; Hohn and Murray, 1977; Hendrix et al., 1983)
Infectious phage particles are obtained in the presence of phage proteins and phage tail components and are used for infecting susceptible bacteria. Clones are selected on the basis of antibiotic resistance and can be amplified subsequently.
Transfection is achieved by incubating a mixture of infectious phage particles obtained by in vitro packaging of recombinant DNA molecules with pretreated sensitive cells. This step is followed by amplification of the library after the titre has been determined.
| Strain | Genotype/Phenotype | Vector | Ref. |
| E. coli HB101 | F-, hsdS20 (r-B, m-B), supE44, ara-14, galK2, lacY1, proA2, rpsL20 (strR), xyl-5, mtl-1, recA13 | cos | 1) |
| E. coli ED8767 | recA56, supE, supF, hsds-R+M+, mer- | cos | 2) |
| E. coli 1400 | cos | 3) | |
| E. coli C600 | F-, supE44, thi-1, thr-1, leuB6, lacY1, tonA21, λ- | 4) | |
| E. coli Y1090 | Δ lacU169, proA+, Δlon, araD139, strA, supF(trpC22::Tn10) | λgt11 | 5) |
| E. coli BNN102 | hsdR-, hsdM+, supE, thr, leu, thi, lacY1, tonA21, hf1A150(chr::Tn10) | λgt10 | 6) |
| E. coli Q358/Q359 | hsd(r-K, m+K), supF, Φ80R hsd(r-K, m+K), supF, Φ80R, P2 | λ2001 λ1059 | 7) |
| E. coli 490A | hsdS-, mer-, thi-, leu-, lac, recA-, (thr) | cos | 8) |
| E. coli K803 | metD, hsdS-nR+, M+, supE, supF, trpR, tonA | cos | 9) |
| E. coli DH5 | cos | 10) | |
| E. coli LE392 | F-, hsdR514(r-K, m+K), supE44, supF58, Δ (lacIZY)6, galK2, galT22, metB1, trpR55, λ- | Charon 4A | 11) |
| E. coli SF8 | C600thr-, leu-, thi-, r-, m-, recB-, recC-, lop-11, lig+ | cos | 12) |
1) Boyer and Roulland-Dussoix, 1969;
2) Lund, Grosveld and Flavell, 1982;
3) Cami and Kourilsky, 1978;
4) Appleyard, 1954;
5) Huynh, Young and Davis, 1985;
6) Young and Davis, 1985;
7) Karn et al., 1984;
8) Karn et al., 1980;
9) Buckel and Zehelein, 1981;
10) Wood, 1966;
11) Murray, Brammer and Murray, 1977;
12) Cameron et al.,1975
Table 6: Strains of Escherichia coli Used for Cloning in λ and Cosmid Vectors
Depending upon the vector used (λ or cosmid) the established gene library can be stored as a suspension culture at 4°C for several years or as a glycerol stock at -80°C for at least one year.
The starting material for the establishment of a cDNA library is mRNA isolated either from pieces of tissue or whole organs. cDNA libraries are not complete collections of the entire genetic information of an organism. Instead they represent the current state of gene expression in tissues or organs. It is therefore critical to keep in mind the current state of activity of a particular organ or tissue when cDNA libraries are prepared. A decisive step in cDNA synthesis is the formation of a double-standed DNA copy of the mRNA. This requires the starting mRNA to be of the highest possible quality. The isolated total RNA contains considerable amounts of ribosomal RNA (rRNA) and transfer RNA (tRNA) while the proportion of messenger RNA (mRNA) may be comparatively low.
Most mRNAs are characterized by a polyadenylated 3 end which is known as poly(A) tail. This sequence is lacking in other structural RNAs. Separation of mRNA from other RNA species can be achieved by poly(A)+ selection. Total RNA is first heat-denatured to remove any secondary structures and subsequently passed over an oligo(dT) cellulose column (Aviv and Leder, 1972). At high salt concentrations RNA containing poly(A) ends will bind to oligo(dT) cellulose while RNA without tails will pass through the column. Polyadenylated RNA can then be eluted from the column by removing the salt which destabilizes the dT:rA hybrids. Column chromatography is repeated several times to reduce contamination with other RNA species.
Another protocol involves the use of poly(U) sepharose instead of oligo(dT) (Moore and Sharp, 1984). The advantage of poly(U) sepharose over oligo(dT) cellulose is that it binds longer stretches of nucleotides and that liquid passes much quicker through the column.
As a rule of thumb one may keep in mind that approximately 1 percent of the input RNA should be recoverable as poly (A)+ RNA. This mRNA should produce a smear during gel electrophoresis with sizes starting at approximately 20 kb and becoming progressively smaller. Highest intensities should be found in a region between 5 and 10 kb.
The first strand of the cDNA is synthesized enzymatically by reverse transcriptase with poly(A)+-mRNA as starting material. The second strand and hence the finished cDNA product is synthesized by a combination of treatments with RNase H, DNA polymerase I and E. coli ligase. Several different protocols have been developed for the synthesis of the second strand during the years (Okayama and Berg, 1982; Gubler and Hoffman, 1983; Neve et al., 1986).
In another reaction step the cDNA is treated in a way allowing its ligation with suitable vector DNA. The following steps described below are required for establishing a genomic DNA library.
The isolation of genes or coding regions of a gene (cDNA) is usually achieved by screening a gene library.
It is possible to screen many recombinant clones simultaneously by growing individual clones of a gene library on agar plates and transferring them to nitrocellulose filters (Grunstein and Hogness, 1975; Benton and Davis, 1977; Cami and Kourilsky, 1978).
A critical parameter governing the screening process is the number of clones that must be analysed to find a particular gene. This number should be calculated on the basis of theoretical considerations before the experiment is carried out. One problem that frequently occurs is the selective loss of certain clones and the over-representation of other clones after amplification of the library. If a desired clone cannot be detected in the library it will be necessary to screen another library that has been established independently.
Particular clones can be identified, for example, by hybridization with suitable probes. These may be either isolated genes, gene fragments derived from the same or other species, or oligonucleotides produced synthetically. Probes are usually labeled by the nick translation procedure (Rigby et al., 1977), random-priming technique (Feinberg and Vogelstein, 1983, 1984), or by endlabelling with radioactive nucleotides. Newly developed techniques allow fusion products (either proteins or mRNA) to be identified with suitable antibodies after cloning in expression vectors (e.g. lgt11). This process is also known as immuno-screening (Erlich, Cohen and McDevitt, 1978; Helfman, et al., 1983; Huyhn, Young and Davis, 1985).
Repetitive screening of a library should yield individual clones which hybridize or react specifically and reproducibly with related probes. These clones are subsequently isolated and amplified and yield recombinant DNA for further investigation. If isolated clones contain very large DNA inserts - as may be the case with cosmid libraries - they are fragmented by cleavage with restriction endonucleases and subcloned in plasmid vectors such as pUC vectors. This process can also be combined with the establishment of a physical gene map (Smith and Birnstiel, 1976).
Depending upon the availability of data concerning the structure of other related genes, some information about homologies and the approximate organization of the clone may be obtained. Further information is obtained by determining the DNA sequences of individual overlapping subclones (Maxam and Gilbert, 1980; Sanger, Micklen and Coulson, 1977). These sequence data can be processed by means of suitable computer programmes. Eventually one contiguous stretch of sequence is obtained from individual subfragments, thus completing the full characterization of the isolated clone. Further experiments are required to decide whether the isolated clone contains a specific gene or not. Some of the theoretical considerations and experimental approaches for this type of analysis will be discussed briefly below:
Search for homologies with other known genes on the nucleotide level. A number of gene data-banks such as EMBO and Microgenie contain entries of all known and sequenced nucleic acids and proteins derived from prokaryotes and eukaryotes. The sequence of the clone under study may be compared with those stored in these data banks.
Comparison of homologies between known proteins whose sequence and structure is known with amino acid sequence information predicted from the DNA sequence of the clone under study.
Search for structural elements such as start and stop codons, promoter elements, splice signals, polyadenylation signals, enhancer elements and protein binding sequences.
Hybridization experiments with known genes or synthesized oligonucleotides as probes (Southern and Northern blotting experiments).
Detection of specific proteins synthesized by in vitro translation techniques, e.g. by Western blotting analysis.
Transfection and expression in tissue culture.
Promoter assays using deletion constructs (CAT assay, Laimins et al., 1982).
Studies involving nuclease S1, exonuclease III (S1 protection assay, S1 mapping, intron mapping) and primer extension experiments (Berg and Sharp, 1977; Weaver and Weissmann, 1979).
Once a gene has been studied in terms of DNA, RNA and protein structures, short or long-term storage is possible either as cosmid, λ, or plasmid clones in suspension culture (4°C), glycerol culture (-80°C), on agar plates (4°C), or as aqueous DNA solution (4°C, -20°C).
The development of molecular biological techniques for the establishment of gene libraries has allowed a great number of genes of viruses, bacteria, yeasts, reptiles, mammals, and higher eukaryotes such as plants, fish, and birds to be isolated, characterized and classified.
All known nucleotide and protein sequences are stored in special data bases which are updated annually. At present there are three important data banks:
Genebank (US government)
European Molecular Biology Laboratory (EMBL) Data Library
National Biomedical Research Foundation (NBRF) Nucleic Acid Database.
The NBRF data base also contains the most important and most comprehensive collection of protein sequences.
Apart from these huge data bases there are a number of computer analysis programmes which may be used with personal computers such as Macintosh or IBM computers. They include programmes such as FASTN and FASTP (Lippman and Pearson, 1985), ANALYSEQ (Staden, 1986), Microgenie (Korn and Queen, 1984) which can be used with IBM-compatible systems, and DNA Inspector II, MacMolly (Soft-Gene, Berlin) DNA-Strider, GELREAD, GELSIZE, MAP, COMPLEMENT, which run on Apple's Macintosh family. Entire volumes of the journal, Nucleic Acids Research, describing these and other programmes are usually issued once a year.
Data bases provide information allowing determination of the structure of genes and the subsequent chemical synthesis of oligonucleotides which may be used to detect other genes (Caruthers, 1985). Chemical synthesis not only provides means to obtain short oligonucleotides but can also be used to synthesize complete genes. This has been demonstrated, for example, by Itakura et al. (1977) who synthesized the somatostatin gene. They have ligated eight different oligonucleotides named A-H to piece together the entire coding sequence of somatostatin and have been able to express this gene under the control of the lac system in E. coli. Other genes such as the A and B chains of human insulin (Crea et al., 1978), human leukocyte interferon (Edge et al., 1981), human proinsulin (Brousseau et al., 1982) have also been synthesized chemically. The different strategies of gene synthesis have been reviewed recently (Gassen and Lang, 1982); Mizuno, 1986; Engels and Uhlmann, 1988).
The technique of chemical DNA synthesis opens up the possibility of synthesizing any conceivable gene. Experiments carried out predominantly with transgenic mice have demonstrated that the biological activity of proteins (e.g. hormones) produced from a transplanted heterologous gene can be retained in many cases. It should therefore be possible to produce transgenic cattle by resorting to the use of heterologous genes coming from other species if homologous genes are either unknown or not available.
As described in Section 3.1.2 the starting material for establishing a library is usually high-molecular weight genomic DNA or poly (A)-selected tissue-specific mRNA. In addition, isolated chromosomes may also be used for preparing chromosome-specific DNA libraries.
Liver tissue is commonly used as a source of DNA for preparing genomic libraries. This tissue can be obtained from biopsy material, or directly after slaughtering and can be stored without considerable concomitant degradation of the DNA either at -20 °C or at -80 °C. For the generation of cosmid libraries it will be advisable to cool the entire tissue as quickly as possible to inactivate endogenous nucleases. If this is impossible the DNA should be isolated immediately. Isolated genomic DNA can then be stored in DNA storage buffer (10 mM Tris-HCl, pH 7.5; 1 mM EDTA) for prolonged periods of time. The DNA can also be stored at -20°C in small batches. Repeated freezing and thawing cycles may lead to DNA degradation.
If a cDNA library is established the fresh tissue must be reduced to pieces which should be stored immediately at -192°C in liquid nitrogen unless one wants to run the risk of obtaining fragmented mRNA. Storage at -20°C or at -80°C is not possible. However, isolated mRNA may be stored in ethanol at -20°C for indefinite periods.
The establishment of chromosome-specific libraries follows the same rules governing the isolation of high-molecular weight DNA. In addition, the special conditions and rules for chromosome isolation must be closely followed.
Direct microinjection of DNA has become a widely used technique for gene transfer. Gordon et al. (1980) were the first to show that it is possible to microinject recombinant plasmids into mouse oocytes and that the injected DNA can be detected in a fraction of the fully developed mice by Southern blotting techniques. Additional experiments have demonstrated that this DNA had been integrated into the genome (Gordon and Ruddle, 1981; Wagner, Stewart and Mintz, 1981; Wagner et al., 1981; Brinster et al., 1981; Costantini and Lacy, 1981). Several reports have also described expression of the newly integrated DNA.
Gene transfer is aimed at creating organisms in which as many cells of an individual as possible will carry the new gene. From this it follows that the transfer of the new gene into the organisms must be undertaken very early in its development. Microinjection of DNA is virtually carried out either with fertilized oocytes or, at most, with 2-cell stages.
A programme for the production of transgenic cattle can be subdivided into five main phases:
Preparation of donor animals, isolation of fertilized oocytes, and visualization of pronuclei
Preparation of DNA solution for injection
Microinjection of DNA solution into pronuclei
Transfer of injected zygotes into recipients
Screening of animals born for integration of the transferred gene.
In cattle the isolation of fertilized oocytes is an extremely laborious process because no non-surgical techniques are available. It requires surgical opening of the abdominal cavity by making an incision in the median line, which allows flushing of the oviducts. Surgery is also associated with great expense in terms of personnel and technical supply. An alternative is to obtain embryos from slaughtered superovulated animals. Reproductive organs may be obtained easily and quickly during the normal process of slaughtering. Oocytes are isolated by flushing the oviduct approximately 12 to 24 hours after fertilization. They are washed several times and can be kept in standard culture medium.
In cattle, but also in some other domesticated mammals, the pronuclei in fertilized oocytes are invisible because they are covered by lipid-containing dark granulae. They can be visualized by centrifugation of the oocytes at 15000 g for three minutes. During centrifugation granulae will migrate to one pole of the oocyte, while the pronuclei will remain in position in the middle of the oocyte cell. Separation of cellular components allows visualization of the pronuclei (Wall et al., 1985). The gene to be injected is precipitated, washed and dissolved in injection buffer. All solutions used for DNA injections must be filtered sterile to remove any contaminating particles, because impure preparations of DNA may obstruct the injection pipette. The concentration of the DNA is adjusted to obtain approximately several hundred copies of the gene to be injected per picolitre.
The most suitable set-up for carrying out microinjections consists of an inverted microscope and two Leitz micromanipulators (Figure 20). A microscope slide containing one drop of culture medium which is covered with paraffin oil is placed under the microscope. The manipulator at the left-hand side holds the holding pipette, while the injection pipette is fastened to the other manipulator. The injection pipette is filled with DNA solution and is then connected by a silicon hose to the injection apparatus. The oocytes to be injected are placed in the drop of culture medium, held by the holding pipette and turned, if necessary, until the pronuclei are in the correct position. The injection pipette is then used to penetrate the zona pellucida, the cellular membrane, and the membrane of one of the pronuclei and to inject the DNA (Figure 21). Successful injection is indicated by an increase in volume of the pronucleus. Following injection the oocytes are transferred into a culture dish, where they can be inspected after a short time to select degenerated oocytes.
Surviving zygotes are transferred into the oviducts of synchronized recipients, for which surgery is mandatory. A possible simplification of bovine gene transfer programmes is the fact that injected oocytes may be cultivated in vivo for several days, using the oviducts of pseudo-pregnant rabbits, sheep, pigs, or cattle.
Embryos can be recovered after a period of approximately six days by flushing the uterus of these intermediate recipients and can be evaluated with respect to their development. Normally developed bovine embryos (morulae - blastocysts) may then be transferred to synchronized recipients by conventional non-surgical techniques.

Figure 20: Set-up for DNA Microinjection into Pronuclei
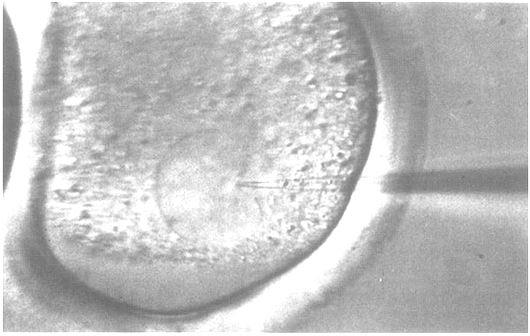
Figure 21: Microinjection into the Pronucleus of a Bovine Zygote
Tissue samples are collected from animals that have developed from microinjected oocytes. Blood samples are best suited for calves. Leukocytes which contain nuclei, are a good source of DNA which can be tested by Southern blot analysis for the presence of injected DNA.
Mann, Mulligan and Baltimore (1983) have developed a cell line, termed Ψ-2 packaging cell line, which packages defective recombinant viral genomes into viable virus particles without requiring helper viruses. The virus particles containing the defective genome are capable of infecting cells and of integrating their genome into the cellular genome. Since the defective genome does not possess all genes required for the creation of intact virions, the host cell does not produce new virus particles.
Stuhlmann, Jähner and Jaenish (1981) have injected cell line Ψ-2–2–5 together with M-MuLV-infected cells into the blastocoele of mouse embryos and have transferred them into acceptor mice. The genome of the replication-defective murine sarcoma virus which carried the Eco-gpt marker gene (MSV-gpt) was packaged within the cell line Ψ-2–2–5. Injection into the inner cells mass of the embryos with MSV-gpt resulted in an infection. More than 50 percent of the animals born displayed a M-MuLV viraemia. In addition expression of the MSV-gpt genome was observed in a variety of tissues in 25–30 percent of these animals. This result demonstrated that retroviral vectors can be used for the introduction of foreign genes into mammals. Van der Putten et al. (1985) have created transgenic mice by cultivating 8-cell stage embryos with digested zona pellucida on a layer of Ψ-2 cells. This cocultivation resulted in an infection of the embryos with retroviral recombinant genomes, which are packaged by Ψ-2 cells.
Jaenisch and Mintz (1974) have injected purified SV40 DNA into the blastocoele of mouse embryos; 40 percent of these blastocysts developed into live tumour-free animals after transfer into suitable recipients while 40 percent revealed the presence of some integrated SV40 DNA genomic elements in some tissues. These animals had therefore incorporated SV40 DNA in their somatic cells and could be regarded as genetic mosaics.
One way to obtain transgenic bovine individuals would be the generation of suitable chimaeras although it must be said that until now some of the techniques required have not been used successfully with cattle.
In mice it has been possible to induce teratomas by ectopic implantation of embryos (day 3–6) into testicular tissue (Stevens, 1968). These tumours were indistinguishable from spontaneously arising, transplantable teratomas (Stevens, 1970). Some of the transferred embryos retained undifferentiated cells for a long time. Upon intraperitoneal injection of these teratomas, embroid bodies were obtained which morphologically resembled mouse embryos (EC-cells). Sherman (1975) has reported the cultivation of cells derived from mouse blastocysts which did not give rise to totipotent cell lines. Evans and Kaufmann (1981) were the first to isolate totipotent cell lines directly from mouse embryos cultivated in vitro (EK-cells). Martin (1981) has isolated totipotent cell lines by conditioning the culture medium of blastocysts with medium in which teratocarcinoma stem cells had been grown previously. In female animals of mouse strain LT/SV teratocarcinomas developed in the gonads of parthogenetically activated oocytes (Stevens and Varnum, 1974).
Since spontaneous teratomas are rare in cattle (Dahme and Weiss, 1983; Thiel and Weingärtner, 1984) suitable cells lines could be obtained by ectopic implantation in vivo or by long-term in vitro culture of blastocysts. Voelkel et al. (1984) have succeeded in cultivating bovine blastocysts in vitro from day 7 to day 17.5.
Embryonal teratocarcinomas injected into the blastocoele are incorporated into the embryo during development. This gives rise to chimaeras. The injection of a single cell into a blastocyst may also give rise to chimaeras.
The integration of embryonal teratocarcinoma cells into the germ line of chimaeras has been demonstrated by several authors (Mintz and Illmensee, 1975; Illmensee, 1978; Stewart and Mintz, 1981; Bradley et al., 1984). Test crosses of chimaeric animals have revealed the presence of the teratocarcinoma genome in subsequent generations. Mintz (1977, 1979) has suggested the possibility of using teratocarcinoma cells as vehicles allowing introduction of mutated genes into mice.
Pellicer et al. (1980) have transferred genes into teratocarcinoma cells by Calcium phosphate treatment. The genes were integrated and were also expressed. The cells retained the new genes even after long-term in vivo passage of the tumours. This approach appears to be promising for generating transgenic animals. Another possibility is demonstrated in Figure 22.
The first step is to generate EC or EK cell lines. Donors of embryos are bull dams mated with bull sires. This approach guarantees the generation of genotypes in the cell lines which correspond with the genetic level of test bulls. Only cell lines derived from male animals are used. In Vitro cultured cell lines are then treated with cloned DNA to integrate the desired gene(s). Integration and expression of the new genes are tested during the period of in vitro cell culture.
Embryos containing a marker gene are recovered as morulae or blastocysts from cow dams and are used to produce chimaeric embryos either by aggregation with, or by injection of transgenic EC or EK cells. These chimaeric embryos are transferred to synchronized recipients by standard non-surgical techniques. The calves born are tested whether they are germ line chimaeras. Chimaeras producing semen derived from the transgenic cell line are used for artificial insemination. The resulting calves are paternal half-siblings if they are derived from EC or EK cells. They are hemizygous for the transferred gene.
One of the advantages of the production of transgenic cattle by means of chimeras obtained by embryo manipulation is technical in nature: instead of obtaining and transferring zygotes by surgical techniques, it is possible to use conventional embryo transfer protocols involving non-surgical methods. DNA transfer into cell lines can be achieved by relatively simple methods. Since no embryos are required at this step, the transfer can be repeated as often as necessary until a suitable integration site is found and the expression of the new DNA has been demonstrated. With cell lines an expression test is much simpler to perform and also much cheaper than with animals. It is still an open question whether expression of genes observed in tissue-culture cells will also be observed in the animal. The probability of obtaining the expected germ line chimaeras in cattle is still unknown. This figure will be known as soon as a larger number of chimaeras have been produced.
Proof of the stable integration and expression in a host animal of a gene construct introduced by microinjection techniques is obtained in three steps:
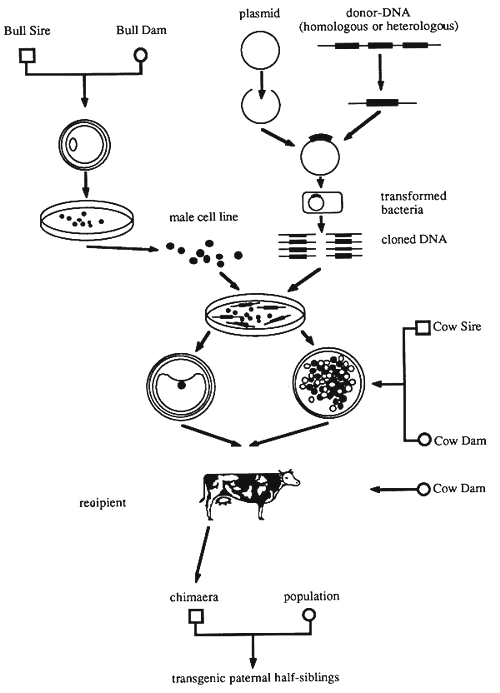
Figure 22: Production of Transgenic Cattle by Production of Chimaeras Derived from Embryos and Embryonal Stem Cells.
1. Detection of the microinjected gene construct in the DNA of the host animal by Southern blot analysis.
Once an animal has been born, DNA is isolated from tissue or blood specimens. It is not necessary to obtain specific high-molecular weight DNA as it is required for establishing genomic libraries. A mean molecular weight of 1.65 × 107 is quite sufficient for the detection of transgenes. Suitable protocols have been worked out by Hogan, Costantini and Lacy (1986).
The isolated DNA is digested to completion with restriction endonucleases and subjected to agarose gel electrophoresis to separate the individual DNA fragments. They are transferred to nitrocellulose or nylon membranes (Figure 23). The filters can be hybridized with radioactively labeled probes (Southern 1975; Meinkoth and Wahl, 1984; Reed and Mann 1985; Wahl, Stern and Stark, 1979). Hybridized filters can be exposed on X-ray film at -80 °C in film cassettes containing intensifier screens (Figure 24). An alternative to the use of radioactive labels is the use of biotinylated dUTP (Langer, Waldroop and Ward, 1981) to label individual DNA fragments. Biotin has a very high affinity towards avidin, which is itself coupled with suitable indicators such as antibodies, enzymes, or dyes. This technique allows minute amounts of biotin/DNA complexes to be detected. If Southern blot analysis reveals the presence and integration of foreign DNA the next step is to determine whether the gene construct is correctly expressed.
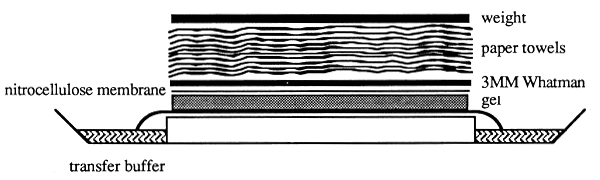
Figure 23: Southern Blot Analysis (Southern, 1975)
DNA fragments separated by agarose gel electrophoresis are subjected to alkaline denaturation and are then
transferred by capillary forces from the gel to a nitrocellulose or nylon membrane placed directly on top of
the gel. Transfer buffer (20×SSC) is sucked through the gel and the membrane by placing approximately
10 cm of paper towels on top of the transfer membrane. A weight which is put on top of the paper towels
ensures equal transfer. Genomic DNA requires approximately 10–16 hours to be transferred completely.
2. Isolation of cytoplasmic RNA and Northern blot analysis.
The tissue specificity of the expression of a gene or gene construct is determined, among others, by its promoter. If correct transcription of a microinjected gene construct is to be analysed RNA is therefore isolated from tissues in which the highest degree of expression is to be expected. The RNA is prepared according to protocols described by Chirgwin et al. (1979), or Glisin, Crkvenjakov and Byus (1974) which are based on cell lysis and denaturation of proteins by guanidinium isothiocyanate.
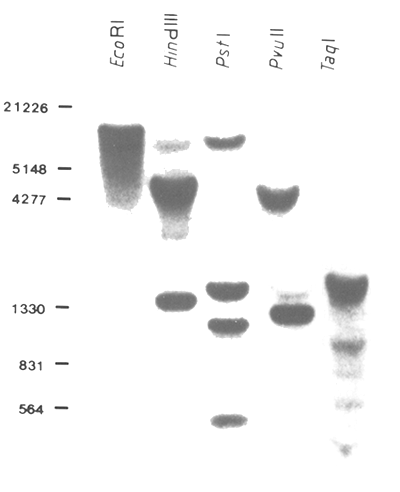
Figure 24: Autoradiography of a Southern Blot Filter
DNA of a transgenic pig was isolated from the terminal part of the tail and electrophoresed on an agarose
gel after restriction with the indicated endonucleases. After transfer to a nitrocellulose filter the DNA was
hybridized with a radioactive-labelled DNA probe. The Southern filter was washed and exposed at -80°C
on an X-ray film for 24 hrs. Sizes of λ marker are shown on the left-hand side in bp.
The RNA is subsequently electrophoresed in denaturing agarose formaldehyde gels, or treated with glyoxal and dimethylsulfoxide (DMSO) and is then transferred to nitrocellulose or nylon filters similar to the process described for DNA in Southern blot analysis (Figure 25) (Thomas, 1980).
Ribonuclease or primer extension experiments are carried out to obtain information on the amount, structure, size, and rate of synthesis of RNA S1 nuclease. Single-stranded probes which are complementary to the RNA to be analysed are used for S1 analysis (Sharp, Berk and Berget, 1980) and ribonuclease protection assays (Chamberlin and Ryan, 1982). These techniques allow 5' and 3' ends and the amounts of specific RNAs to be determined. S1 analysis employs endlabeled single-stranded DNA probes while labeled RNAs obtained by in vitro transcription mediated by SP6, T7, and T3 RNA polymerase are used in ribonuclease protection assays (Kassevetis et al., 1982; Melton et al., 1984). Primer extension analysis requires radioactively labeled oligonucleotides to be hybridized to RNA and subsequent synthesis of copies of the template RNA by reverse transcription. Synthesis products can be analysed on polyacrylamide sequencing gels (McKnight and Kingsbury, 1982; Jones, Yamamoto and Tjian, 1985).
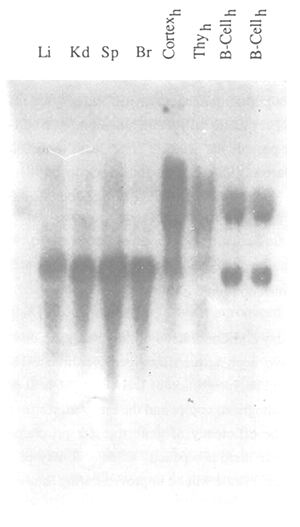
Figure 25: Autoradiography of a Northern Blot Filter
RNA was isolated from different tissues according to Chirgwin et al. (1979) and electrophoresed on a
denaturing 1.2 percent formaldehyde agarose gel. The RNA was transferred to a nylon membrane and
hybridized with a radioactive-labelled DNA probe. After hybridization the filter was washed and exposed to
an X-ray film at -80°C for 24 hrs. Li: liver; Kd: Kidney; Sp: spleen; Br: brain; Cortexh: human cortex;
Thyh: human thymus; B-Cellh: human B-cell line.
All three techniques allow analysis of the size and amount of specific RNAs. Primer extension analysis differs in one decisive point from the other two methods. Reverse transcriptase yields a complete copy of RNA irrespective of the presence of any splice sites. Primer extension analysis therefore complements S1 analysis because it allows exact determination of the 5' ends of transcripts.
3. Translation studies
If correct transcription of a microinjected gene construct is demonstrated it is also necessary to analyse translation. This is usually done by determining first whether a translation product is formed and if so by measuring the amounts of protein. Several colorimetric tests can be used to quantitate proteins (Bradford technique, Lowry technique). They can also be analysed by gel electrophoretic methods and subsequent staining with Coomassie blue or silver stain. Another technique is transfer of proteins to nitrocellulose or nylon filters and subsequent Western blot analysis. Clinically useful techniques such as radioimmunoassay, ELISA (enzyme linked immunosorbent assay) may also be used. They are usually available as kits. We will not discuss these techniques in detail but rather provide the necessary references (Bradford, 1976; Lowry et al., 1951; Peterson, 1979; Darbre, 1986; Hames and Rickwood, 1981; Oakley, Kirsch and Morris, 1980; Moeremans, Daneels and De Mey, 1985; Moeremans et al., 1986; Salinovich and Montelaro, 1986; Peterson, 1977).
Animals which carry additional newly integrated gene constructs produced by in vitro recombination have been termed transgenic animals. The integrated foreign DNA construct is a transgene. Integration of DNA into the host genome is a random process, i.e. the number of integrated copies and the integration sites cannot be determined before the experiment. The efficiency of gene transfer programmes is generally very low. Especially with cattle there is a paucity of data. It may be assumed however that gene transfer programmes in cattle will be improved during future years.
Little is known about the processes taking place during the integration of foreign DNA into the host genome. It is also not known when integration occurs and whether integration is stable during the first divisions of an embryo. It has been established, however, that mosaics may be obtained in spite of DNA being injected into the pronuclei of a zygote. Mosaics are animals which consist of different cell lines, although all of them derived from the same zygote.
Transgenic mosaics are composed of cells which harbour the transgene and those that do not. This creates problems when breeding lines and transgenic offspring are to be produced. If the cells of the gonads do not contain transgenes, they may not be passed on to the offspring. Based on experience gathered from experimental animals one may deduce that approximately one third of all transgenic animals generated by microinjection will be mosaics which do not pass on the transgene. At least with respect to permanent reactivation of a desired gene these animals are useless because the transgenic trait is only present for the life span of G0 animals.
Transgenic cattle which harbour the gene construct in their germ cells may pass on the transgene to the offspring. These genes are normally inherited as Mendelian genes. Under normal circumstances integration occurs only at one site within a chromosome. These primary transgenic animals have therefore been termed hemizygous. By definition they are not heterozygous because the transgenic allele is missing from the homologous chromosome. Statistically approximately 50 percent of the offspring of a transgenic animal whose gonads are not mosaics will inherit the transgene.
If the gonads contain transgenic and non-transgenic cell lines the frequency of transgenic offspring may be between 0–50 percent, depending upon the degree to which the transgenic cell lines contribute to the composition of the gonads. This problem is only encountered in the offspring derived from the primary transgenic animal. If a transgenic G1 generation has been obtained, the transgenic offspring will harbour the gene construct in all its somatic and germ cells. The only uncertain factor is introduced by the instability of the integrated transgene. Some cases have been described in which the transgene was lost. It has been assumed that the integrated foreign DNA may be removed from the genome by repair mechanisms. In some cases this may lead to offspring, none of which would be trans-genic.
There are relatively few exceptions of animals containing more than one integration site for a transgene. If the distance between these sites is great enough to allow random recombination more transgenic offspring may be generated. A transgenic animal harbouring two independent integration sites would transmit the transgene to 75 percent of its offspring; 25 percent of these would inherit both transgenes, 25 percent would harbour one or other of the two integration sites and 25 percent of the offspring would not be transgenic.
By mating hemizygous transgenic animals one would normally obtain 25 percent homozygous transgenic, 50 percent hemizygous transgenic, and 25 percent normal offspring. Since integration is essentially a random event one might also expect cases in which the transgene has been positioned in the vicinity of a gene which is essential for foetal development. No problems should be encountered as long as an intact allele is retained functional on the homologous chromosome. An exception would be additive genetic effects if integration occurred in one of these genes, thus leading to a reduced expression of its allele.
No transgenic offspring are to be expected in matings between hemizygous animals carrying an insertion mutation if the gene that has been affected by the integration event is essential for further development of the foetus. Wagner et al. (1983) have observed this case in the offspring of transgenic mice carrying the hGH gene. They have described two lines of transgenic animals characterized by prenatal mortality of homozygous animals due to an insertion of the gene construct into an essential DNA region.
| Generation | Technique | Mating | Homozygosity test |
| 0 | primary transgenes × animals from population T0 × 00 | ||
| 1 | Progeny test | 50% T0 half-siblings T0 bulls × 00 | inbreeding T0 × T0 |
| 2 | Propagation and enlargement of genetic basis | 50% T0 cousins Inbreeding T0 × T0 | 25% TT Vitality test |
| 3 | Establishment of pure homozygous lines inbreeding rate 3% | 25% TT Selection |
Table 7: Breeding Protocol and Test Procedure for Primary Transgenic Animals (According to Smith, Meuwissen and Gibson, 1987).
Hemizygous lines were able to be bred by mating with normal mice and led to 50 percent healthy transgenic offspring.
Woychik et al. (1985) have found and insertion mutation in transgenic mice which led to the well-known defect of malformation of the limbs. In this case the transgene obviously integrated into the gene(s) required for limb development; 21 percent of the offspring (n = 186) obtained by mating hemizygous parents did not display any signs of malformation.
A breeding protocol for testing transgenic animals and establishing homozygous lines has been described by Smith, Meuwissen and Gibson (1987) (Table 7).
The practical conclusions that can be drawn from these observations are that it will not be sufficient to produce one transgenic animal only if the aim is to introduce a certain gene into a cattle population. A part from quickly creating problems through inbreeding, the chances of establishing a functional transgenic line with a single integration of a transgene would be too low.
The following is a brief summary of points that must be expected from a transgenic breeding line:
Taking into consideration experiences gathered so far it can be estimated that approximately five transgenic animals will have to be produced under normal conditions in order to guarantee with sufficient probability the generation of a stable homozygous transgenic line displaying good expression of the transgene and possessing the expected phenotype. If one takes into consideration the efficiency with which transgenic cattle can be produced also by DNA microinjection (i.e. 0.5 percent of transgenic cattle per injected oocyte) it will become clear that reactivation of a single conserved gene in a future population is associated with considerable expense and effort.
![]()
![]()
![]()