![]()
![]()
![]()
(At Step 3 of the Procedure)
FOREWORD
The Guidelines are intended to assist in ensuring the reliability of analytical results in checking compliance with maximum residue limits of foods moving in international trade. Reliable analytical results are essential to protect the health of consumers and to facilitate international trade.
In addition to the present Guidelines, other relevant Codex recommendations elaborated by the Codex Committee on Pesticide Residues in the field of enforcement of Codex maximum limits for pesticide residues are as follows:
1 Recommended Method of Sampling for the Determination of Pesticide Residues (ref.: CAC/VOL XIII - Ed.2, Part VI or CAC/PR 5-1984), as amended with respect to meat and poultry (ALINORM 91/40; see also ALINORM 89/24A, Appd. II and ALINORM 91/24A Appd. VIII).CODEX GUIDELINES ON GOOD PRACTICE IN PESTICIDE RESIDUE ANALYSIS2 Portion of Commodities to which Codex Maximum Residue Limits Apply and which should be analysed (ref.: CAC/VOL XIII - Ed. l, Part V or CAC/PR6-1984).
3 Explanatory Notes on Codex Maximum Limits for Pesticide Residues (ref.: CAC/VOL XIII - Ed. 1, Part III).
4 Recommendations for Methods of Analysis of Pesticide Residues (ref.: CAC/VOL XIII Ed. 2 part VIII or CAC/PR 8-1984)
5 Codex Classification of Food and Animal Feed (ref.: CAC/PR4-1989)
1. INTRODUCTION
The Codex document ALINORM 76/24 Appendix IV (Report of the ad hoc Working Group on Methods of Analysis) contained the following statement:
“It was considered that the ultimate goal in fair practice in international trade depended, among other things, on the reliability of analytical results. This in turn, particularly in pesticide residue analysis, depended not only on the availability of reliable analytical methods, but also on the experience of the analyst and on the maintenance of ‘good practice in the analysis of pesticides’.”
These guidelines define such good analytical practice and may be considered in three inter-related parts:
The Analyst (par. 2);The requirements for facilities, management, personnel, quality assurance and quality control, documentation of results and row data, and relevant subjects, which are considered as pre-requisites for obtaining reliable and traceable results are described in general in the ISO/IEC 17025 Standard (1999) and in a series of OECD GLP Guidance Documents, in the corresponding national laws and regulations. This Codex Guidelines, which are not exhaustive, outline the most essential principles and practices to be followed in the analysis of pesticide residues.
Basic Resources (par. 3);
The Analysis (par.4).
2. THE ANALYST
2.1 Residue analysis consists of a chain of procedures, most of which are known, or readily understood, by a trained chemist, but because the analyte concentrations are in the range mg/kg to mg/kg and because the analyses can be challenging, attention to detail is essential. The analyst in charge should have an appropriate professional qualification and be experienced and competent in residue analysis. Staff must be fully trained and experienced in correct use of apparatus and in appropriate laboratory skills. In addition, each analyst using the method for the first time should complete the tests specified in sections 4.4.5 of Table 4 to demonstrate that they can use the method within the expected performance parameters established during method validation prior to applying the method for analysis of samples. They must have an understanding of the principles of pesticide residue analysis and the requirements of Analytical Quality Assurance (AQA) systems. They must understand the purpose of each stage in the method being used, the importance of following the methods exactly as described and of noting any unavoidable deviations. They must also be trained in the evaluation and interpretation of the data that they produce. A record of training and experience must be kept for all members of staff.
2.2 When a laboratory for residue analysis is set up, the staff should spend some of their training period in a well established laboratory where experienced advice and training is available. If the laboratory is to be involved in the analysis for a wide range of pesticide residues, it may be necessary for the staff to gain experience in more than one established laboratory.
3. BASIC RESOURCES
3.1 The Laboratory
3.1.1. The laboratory and its facilities must be designed to allow tasks to be allocated to welldefined areas where maximum safety and minimum chance of contamination of samples prevail. Laboratories should be constructed of and utilise materials resistant to chemicals likely to be used in the area. Under ideal conditions, separate rooms would be designated for sample receipt and storage, for sample preparation, for extraction and clean-up and for instrumentation used in the determinative step. The area used for extraction and clean-up must meet solvent laboratory specifications and all fume extraction facilities must be of high quality. Sample receipt, storage and preparation should be handled in areas devoted to work at residue levels. Maintenance of sample integrity and adequate provisions for personal safety are priority requirements.
3.1.2 Laboratory safety must also be considered in terms of what is essential and what is preferable, as it must be recognised that the stringent working conditions enforced in residue laboratories in some parts of the world could be totally unrealistic in others. No smoking, eating, drinking or application of cosmetics should be permitted in the working area. Only small volumes of solvents should be held in the working area and the bulk of the solvents stored separately, away from the main working area. The use of highly or chronically toxic solvents and reagents should be minimised whenever possible. All waste solvent should be stored safely and disposed of both safely and in an environmentally protective manner taking into account the specific national regulations where available.
3.1.3 The main working area should be designed and equipped for utilisation of an appropriate range of analytical solvents. All equipment such as lights, macerators and refrigerators should be “spark free” or “explosion proof”. Extraction, clean-up and concentration steps should be carried out in a well ventilated area, preferably in fume cupboards.
3.1.4 Safety screens should be used when glassware is used under vacuum or pressure. There should be an ample supply of safety glasses, gloves and other protective clothing, emergency washing facilities and a spillage treatment kit. Adequate fire fighting equipment must be available. Staff must be aware that many pesticides have acutely or chronically toxic properties and therefore, great care is necessary in the handling of standard reference compounds.
3.2 Equipment and Supplies
3.2.1 The laboratory will require adequate, reliable, supplies of electricity and water. Adequate supplies of reagents, solvents, gasses glassware, chromatographic materials, etc., of suitable quality are essential.
3.2.2 Chromatographic equipment, balances, spectrophotometers etc. must be serviced and their performance validated regularly and a record of all servicing/repairs must be maintained for every such item of equipment. Calibration is essential for equipment performing measurements. Calibration curves and comparison with standards may suffice.
3.2.3 Regular calibration and recalibration of measuring equipment should only be done where the possible change in nominal value may significantly contribute to the uncertainty of the measurement. Balances and automated pipettes/dispensers and similar equipment must be calibrated regularly. The operating temperatures of refrigerators and freezers should be checked at specified intervals. All records should be kept.
3.2.4 Although equipment may require periodic updating in order to keep up with developments, the equipment only needs to be sophisticated enough to do the job required.
3.2.5 All laboratories require an adequate range of reference pesticide standards of known and acceptably high purity. The range should cover all parent compounds for which the laboratory is monitoring samples, as well as those metabolites that are included in MRLs.
3.2.6 All analytical standards, stock solutions and reagents must be clearly labelled with an expiry date and stored under proper conditions. ”Pure” reference standards must be kept under conditions that will minimise the rate of degradation, e.g. low temperature, exclusion of moisture, darkness. Equal care must be taken that standard solutions of pesticides are not decomposed by the effect of light or heat during storage or become concentrated owing to solvent evaporation.
4. THE ANALYSIS
The methods applied for the determination of pesticide residues should generally satisfy the criteria given in Table 3.
4.1 Avoidance of contamination
4.1.1 One of the significant areas in which pesticide residue analysis differs significantly from macro-analysis is that of contamination and interference. Trace amounts of contamination in the final samples used for the determination stage of the method can give rise to errors such as false positive or false negative results or to a loss of sensitivity that may prevent the residue from being detected. Contamination may arise from almost anything that is used for, or is associated with, sampling, sample transport and storage, and the analyses. All glassware, reagents, organic solvents and water should be checked for possible interfering contaminants before use, by analysis of a reagent blank.
4.1.2 Polishes, barrier creams, soaps containing germicides, insect sprays, perfumes and cosmetics can give rise to interference problems and are especially significant when an electron-capture detector is being used. There is no real solution to the problem other than to ban their use by staff while in the laboratory.
4.1.3 Lubricants, sealants, plastics, natural and synthetic rubbers, protective gloves, oil from ordinary compressed air lines and manufacturing impurities in thimbles, filter papers and cotton-wool can also give rise to contamination of the final test solution.
4.1.4 Chemical reagents, adsorbents and general laboratory solvents may contain, adsorb or absorb compounds that interfere in the analysis. It may be necessary to purify reagents and adsorbents and it is generally necessary to use re-distilled solvents. De-ionised water is often suspect; re-distilled water is preferable, although in many instances tap water or well water may be satisfactory.
4.1.5 Contamination of glassware, syringes and gas chromatographic columns can arise from contact with previous samples or extracts. All glassware should be cleaned with detergent solution, rinsed thoroughly with distilled (or other clean) water and then rinsed with the solvent to be used. Glassware to be used for trace analysis must be kept separate and must not be used for any other purpose.
4.1.6 Pesticide reference standards should always be stored at a suitable temperature in a room separate from the main residue laboratory. Concentrated analytical standard solutions and extracts should not be kept in the same storage area.
4.1.7 Apparatus containing polyvinylchloride (PVC) should be regarded as suspect and, if shown to be a source of contamination, should not be allowed in the residue laboratory. Other materials containing plasticisers should also be regarded as suspect but PTFE and silicone rubbers are usually acceptable and others may be acceptable in certain circumstances. Sample storage containers can cause contamination and glass bottles with ground glass stoppers may be required. Analytical instrumentation ideally should be housed in a separate room. The nature and importance of contamination can vary according to the type of determination technique used and the level of pesticide residue to be determined. For instance contamination problems which are important with methods based on gaschromatography or high performance liquid chromatography, may well be less significant if a spectrophotometric determination is used, and vice versa. For relatively high levels of residues, the background interference from solvents and other materials may be insignificant in comparison with the amount of residue present. Many problems can be overcome by the use of alternative detectors. If the contaminant does not interfere with the residue determination, its presence may be acceptable.
4.1.8 Residue and formulation analyses must have completely separate laboratory facilities provided. Samples and sample preparation must be kept separate from the all residue laboratory operations in order to preclude cross contamination.
4.2 Reception and storage of samples
4.2.1 Every sample received into the laboratory should be accompanied by information on the sample, on the analysis required and on potential hazards associated with the handling of that sample.
4.2.2 On receipt of a sample it must immediately be assigned a unique sample identification code which should accompany it through all stages of the analysis to the reporting of the results. If possible, the samples should be subject to an appropriate disposal review system and records should be kept.
4.2.3 Sample processing and sub-sampling should be carried out using procedures that have been demonstrated to provide a representative analytical portion and to have no effect on the concentration of residues present.
4.2.4 If samples cannot be analysed immediately but are to be analysed quickly, they should be stored at chill (1-5° C) temperature, away from direct sunlight, and analysed within a few days. However, samples received deep-frozen must be kept at £ - 16° C until unalysis. In some instances, samples may require storage for a longer period before analysis. Storage temperature should be approximately - 20° C, at which temperature enzymic degradation of pesticide residues is usually extremely slow. If prolonged storage is unavoidable, the effects of storage should be checked by analysing fortified samples stored under the same conditions for a similar period. Useful information on storage stability of pesticide residues can be found in the annual publications of FAO titled: Pesticide Residues - Evaluations prepared by the FAO/WHO JMPR, and in the information submitted by the manufacturers for supporting the registration of their pesticides.
4.2.5 When samples are to be frozen it is recommended that analytical test portions be taken prior to freezing in order to minimise the possible effect of water separation as ice crystals during storage. Care must still be taken to ensure that the entire test portion used in the analysis.
4.2.6 Neither the containers used for storage nor their caps or stoppers should allow migration of the analyte(s) into the storage compartment. The containers must not leak. All samples should be labelled clearly with permanent labels and records must be kept. The extracts and final test solution should not be exposed to direct sunlight.
4.3 Standard Operating Procedures (SOPs)
4.3.1 SOPs should be used for all operations. The SOPs should contain full working instructions as well as information on applicability, expected performance, internal quality control (performance verification) requirements and calculation of results. It should also contain information on any hazards arising from the method, from standards or from reagents.
4.3.2 Any deviations from a SOP must be recorded and authorised by the analyst in charge.
4.4 Validation of Methods[28]
4.4.1 An analytical method is the series of procedures from receipt of a sample to the production of the final result. Validation is the process of verifying that a method is fit for the intended purpose. The method may be developed in-house, taken from the literature or otherwise obtained from a third party. The method may then be adapted or modified to match the requirements and capabilities of the laboratory and/or the purpose for which the method will be used. Typically, validation follows completion of the development of a method and it is assumed that requirements such as calibration, system suitability, analyte stability, etc., have been established satisfactorily. When validating and using a method of analysis, measurements must be made within the calibrated range of the detection system used. In general, validation will precede practical application of the method to the analysis of samples but subsequent performance verification is an important continuing aspect of the process. Requirements for performance verification data are a subset of those required for method validation.
Proficiency testing (or other inter-laboratory testing procedures), where practicable, provides an important means for verifying the general accuracy of results generated by a method, and provides information on the between-laboratory variability of the results. However, proficiency testing generally does not address analyte stability or homogeneity and extractability of analytes in the processed sample.
Where uncertainty data are required, this information should incorporate performance verification data and not rely solely on method validation data.
4.4.2 Whenever a laboratory undertakes method development and/or method modification, the effects of analytical variables should be established, e.g. by using ruggedness tests, prior to validation. Rigorous controls must be exercised in respect of all aspects of the method, which may influence the results, such as: sample size; partition volumes; variations in the performance of the clean-up systems used; the stability of reagents or of the derivatives prepared; the effects of light, temperature, solvent and storage on analytes in extracts; the effects of solvent, injector, separation column, mobile phase characteristics (composition and flow-rate), temperature, detection system, co-extractives etc. on the determination system. It is most important that the qualitative and quantitative relationships between the signal measured and the analyte sought is established unequivocally.
4.4.3 Preference should be given to methods having multi residue and or multi matrix applicability. The use of representative analytes or matrices is an important tool in validating methods. For this purpose, commodities should be differentiated sufficiently but not unnecessarily. For example, some products are available in a wide range of minor manufactured variants, or cultivated varieties, or breeds, etc. Generally, though not invariably, a single variant of a particular commodity may be considered to represent others of the same commodity but, for example, a single fruit or vegetable species must not be taken to represent all fruit or vegetables (Table 5). Each case must be considered on its merits but where particular variants within a commodity are known to differ from others in their effects on method performance. Considerable differences in the accuracy and precision of methods, especially with respect to the determination step, may occur from species to species.
4.4.3.1 Where experience shows similar performance of extraction and clean-up between broadly similar commodities/sample matrices, a simplified approach may be adopted for performance validation. A representative commodity may be selected from Table 5 to represent each commodity group having common properties, and used for validation of the procedure or method. In Table 5, the commodities are classified according to the Codex Classification[29].
Some examples of how far the validation data may be extended to other commodities are:
4.4.3.2 Similarly representative analytes may be used to assess the performance of a method. Compounds may be selected to cover physical and chemical properties of analytes that are intended to be determined by the method. The selection of representative analytes should be made based on the purpose and scope of analysis taking into account the following.
(a) The representative analytes selected should:4.4.3 Where appropriate data are already available, it may not be necessary for the analyst to perform all the tests. However, all required information must be included or referred to in the validation records. Table 1 provides an overview of parameters to be assessed for method validation according to the status of the method to be validated. Specific parameters and criteria to be assessed are listed in table 2. Parameters to be assessed should be restricted to those that are appropriate both to the method and to the purpose for which the particular method is to be applied. In many cases, performance characteristics with respect to several parameters may be obtained simultaneously using a single experiment. Test designs where different factors are changed at the same time (factorial experiment designs), may help to minimise the resources required. The performance of the analytical method should be checked, both during its development and during its subsequent use as indicated in section 4.5, according to the criteria given in Table 3.(i) possess sufficiently wide range of physico-chemical properties to include those of represented analytes;(b) As far as practicable, all analytes included in the initial validation process should be those which will have to be tested regularly and which can be determined simultaneously by the determination system used.(ii) be those which are likely to be detected regularly, or for which critical decisions shall be made based on the results.
(c) The concentration of the analytes used to characterise a method should be selected to cover the AL-s of all analytes planned to be sought in all commodities. Therefore the selected representative analytes should include, among others, those which have high and low AL-s. Consequently, the fortification levels used in performance testing with representative analytes/representative commodities may not necessarily correspond to the actual AL-s.
4.4.3.1 Individual (single residue) methods should be fully validated with all analyte(s) and sample materials specified for the purpose, or using sample matrices representative of those to be tested by the laboratory.
4.4.3.2 Group specific methods (GSM) should be validated initially with one or more representative commodities and a minimum of two representative analytes selected from the group.
4.4.3.2 MRMs may be validated with representative commodities and representative analytes.
4.5 Performance verification
4.5.1 The main purposes of performance verification are to:
The results of internal quality control provide essential information on the long term reproducibility and other performance characteristics of the method including the analytes and commodities which were incorporated during the extension of the method.
The basic performance characteristics to be tested and the appropriate test procedures are described in Table 5.
For effective performance verification, analyse samples concurrently with appropriate quality control analyses (blank and recovery determinations, reference materials, etc.). Control charts may be used to check for trends in performance of the method and to ensure that statistical control is maintained.
4.5.2 Construction and use of control charts.
Control chart may be a useful tool for demonstrating the performance of a method and the reproducibility of its selected parameter. One example for that is the control chart for recoveries. Its application depends on the tasks of the laboratory. When large number of the same type of sample is analysed for the same active ingredients the control chart is based on the mean recovery and its standard deviation obtained during the regular use of the method. When small number of each of a large variety of samples are analysed for a great number of analytes with a multi residue procedure the control charts cannot be applied in the usual way. In such cases initially, a control chart is constructed with the average recovery (Q) of representative analytes in representative matrices and the typical within-laboratory reproducibility coefficient of variation (CVAtyp), obtained as described below. When the average recovery data and their coefficient of variation obtained during method validation for individual analyte/sample matrices are not statistically different, each can be considered as an estimate of the true recovery and precision of the method, and with their appropriate combination the typical recovery (Qtyp) and coefficient of variation (CVAtyp) of the method can be established and used for constructing the initial control chart. The warning and action limits are Qtyp ± 2*CVAtyp*Q and Qtyp ± 3*CVAtyp*Q, respectively.
4.5.2.1 When the method is applied for regular analysis of various analyte/matrix combinations represented during the validation of the method, the individual recoveries are plotted on the chart. The reproducibility of the method during its normal use may be somewhat higher then obtained at the validation of the method. Therefore, if some of the recoveries are outside the warning limits or occasionally the action limits, but they are within the ranges calculated from the CVA values specified in Table 3, no special action is required.
4.5.2.2 Based on the additional 15-20 recovery tests performed during the regular use of the method, as part of performance verification, the mean or typical recovery and the CVA shall be recalculated and a new control chart constructed which reflects the long term reproducibility of the application of the method. The new parameters established must be within the acceptable ranges specified in Table 3.
4.5.2.3 If this is not achievable, for example in the case of particularly problematic analytes, results from samples should be reported as having poorer accuracy or precision than is normally associated with pesticide residues determination.
4.5.3.4 During the regular use of the method, if the average of the first ³10 recovery tests for a particular analyte/sample matrix is significantly different (P = 0.05) from the average recovery obtained for the representative analyte/sample matrices, the Qtyp and CVtyp are not applicable. Calculate new warning and action limits for the particular analyte/sample matrix, applying the new average recovery and the CV values measured.
4.5.3.5 If performance verification data repeatedly fall outside the warning limits (1 in 20 measurements outside the limit is acceptable), the application conditions of the method must be checked, the sources of error(s) identified, and the necessary corrective actions taken before use of the method is continued.
4.5.3.6 If performance verification data are outside the refined action limits established according to 4.2.3, the analytical batch involved (or at least samples in which residues found are £ 0.7 AL or 0.5 AL, for regularly and occasionally detected analytes, respectively) should be repeated.
4.5.6.7 Re-analysis of analytical portions of positive samples is another powerful way of performance verification. Their results can be used to calculate the overall within-laboratory reproducibility of the method (CVLtyp) in general or for a particular analyte/sample matrix. In this case, the CVLtyp will also include the uncertainty of sample processing, but will not indicate if the analyte is lost during the process.
4.6 Confirmatory Tests
4.6.1 When analyses are performed for regulatory purposes, it is especially important that confirmatory tests are carried out before reporting adversely on samples containing residues of pesticides that are not normally associated with that commodity, or where MRLs appear to have been exceeded. Samples may contain interfering chemicals that may be misidentified as pesticides. Examples in gaschromatography include the responses of electron-capture detectors to phthalate esters and of phosphorus-selective detectors to compounds containing sulphur and nitrogen. As a first step, the analysis should be repeated using the same method, if only one portion was analyzed initially. This will provide evidence of the repeatability of the result, if the residue is confirmed. It should be noted that the only evidence supporting the absence of detectable residues is provided by the performance verification data.
4.6.2 Confirmatory tests may be quantitative and/or qualitative but, in most cases, both types of information will be required. Particular problems occur when residues must be confirmed at or about the limit of determination but, although it is difficult to quantify residues at this level, it is essential to provide adequate confirmation of both level and identity.
4.6.3 The need for confirmatory tests may depend upon the type of sample or its known history. In some crops or commodities, certain residues are frequently found. For a series of samples of similar origin, which contain residues of the same pesticide, it may be sufficient to confirm the identity of residues in a small proportion of the samples selected randomly. Similarly, when it is known that a particular pesticide has been applied to the sample material there may be little need for confirmation of identity, although a randomly selected results should be confirmed. Where “blank” samples are available, these should be used to check the occurrence of possible interfering substances.
4.6.4 For qualitative confirmation, an alternative technique using different physicochemical properties and/or the use of spectral data is desirable. For quantitative confirmation at least one alternative procedure (which may be a different detection technique, additional ions monitored by mass spectrometry, etc.) should be used. The reported result depends on the methods applied:
If the two results are outside the extreme range the validity of one of them shall be verified and the average of the two conforming results reported.
4.6.5 The necessary steps to positive identification are a matter of judgement on the analyst’s part and particular attention should be paid to the choice of a method that would minimise the effect of interfering compounds. The technique(s) chosen depend(s) upon the availability of suitable apparatus and expertise within the testing laboratory. Some of alternative procedures for confirmation are given in Table 6.
4.7 Mass spectrometry
4.7.1 Residue data obtained using mass spectrometry can represent the most definitive evidence and, where suitable equipment is available, it is the confirmatory technique of choice. The technique can also be used for residue screening purposes. Mass spectrometric determination of residues is usually carried out in conjunction with a chromatographic separation technique to provide retention time, ion mass/charge ratio and ion abundance data simultaneously. The particular separation technique, the mass spectrometer, the interface between them and the range of pesticides to be analysed are usually interdependent and no single combination is suitable for the analysis of all compounds. Quantitative transmission of labile analytes through the chromatographic system and interface is subject to problems similar to those experienced with other detectors. The most definitive confirmation of the presence of a residue is the acquisition of its “complete” electron-impact ionisation mass spectrum (in practice generally from m/z50 to beyond the molecular ion region). The relative abundances of ions in the spectrum and the absence of interfering ions are important considerations in confirming identity. This mode of analysis is one of the least selective and interference from contaminants introduced during the production or storage of extracts should be scrupulously avoided. Mass spectrometer data systems permit underlying interference (e.g. column bleed) signals to be removed by “background subtraction” but this technique must be used with caution. Increased sensitivity can usually be achieved by means of limited mass range scanning or by selected ion monitoring but the smaller the number of ions monitored (especially if these are of low mass), the less definitive are the data produced. Additional confirmation of identity may be obtained (i) by the use of an alternative chromatographic column; (ii) by the use of an alternative ionisation technique (e.g. chemical ionisation); (iii) by monitoring further reaction products of selected ions by tandem mass spectrometry (MS/MS or MSn); or (iv) by monitoring selected ions at increased mass resolution. For quantification, the ions monitored should be those that are the most specific to the analyte, are subject to least interference and provide good signal-to-noise ratios. Mass spectrometric determinations should satisfy similar analytical quality control criteria to those applied to other systems.
4.7.2 Confirmation of residues detected following separation by HPLC is generally more problematic than where gas chromatography is used. If detection is by UV-absorption, production of a complete spectrum can provide good evidence of identity. However, UV spectra of some pesticides are poorly diagnostic, being similar to those produced by many other compounds possessing similar functional groups or structures, and coelution of interfering compounds can create additional problems. UV-absorption data produced at multiple wavelengths may support or refute identification but, in general, they are not sufficiently characteristic on their own. Fluorescence data may be used to support those obtained by UV absorption. LC-MS can provide good supporting evidence but, because the spectra generated are generally very simple, showing little characteristic fragmentation, results produced from LC-MS are unlikely to be definitive. LC-MS/MS is a more powerful technique, combining selectivity with specificity, and often provides good evidence of identity. LC-MS techniques tend to be subject to matrix effects, especially suppression, and therefore confirmation of quantity may require the use of standard addition or isotopicallylabelled standards. Derivatisation may also be used for confirmation of residues detected by HPLC (paragraph 4.6.5.4).
4.7.3 In some instances, confirmation of gas chromatographic findings is most conveniently achieved by TLC. Identification is based on two criteria, Rf value and visualisation reaction. Detection methods based on bioassays (e.g. enzyme, fungi spore, chloroplast inhibition) are especially suitable for qualitative confirmation as they are specific to certain type of compounds, sensitive and normally very little affected by the co-extracts. The scientific literature contains numerous references to the technique, the IUPAC Report on Pesticides (13) (Bátora, V., Vitorovic, S.Y., Thier, H.-P. and Klisenko, M.A.; Pure & Appl. Chem., 53, 1039-1049 (1981)) reviews the technique and serves as a convenient introduction. The quantitative aspects of thin-layer chromatography are, however, limited. A further extension of this technique involves the removal of the area on the plate corresponding to the Rf of the compound of interest followed by elution from the layer material and further chemical or physical confirmatory analysis. A solution of the standard pesticide should always be spotted on the plate alongside the sample extract to obviate any problems of non-repeatability of Rf. Overspotting of extract with standard pesticide can also give useful information. The advantages of thin layer chromatography are speed, low cost and applicability to heat sensitive materials; disadvantages include (usually) lower sensitivity and separation power than instrumental chromatographic detection techniques and need for more efficient cleanup in case of detections based on chemicals colour reactions.
4.8 Derivatisation
This area of confirmation may be considered under three broad headings:
(a) Chemical reactionsSmall scale chemical reactions resulting in degradation, addition or condensation products of pesticides, followed by re-examination of the products by chromatographic techniques, have frequently been used. The reactions result in products possessing different retention times and/or detector response from those of the parent compound. A sample of standard pesticide should be treated alongside the suspected residue so that the results from each maybe directly compared. A fortified extract should also be included to prove that the reaction has proceeded in the presence of sample material. Interference may occur where derivatives are detected by means of properties of the derivatising reagent. A review of chemical reactions which have been used for confirmatory purposes has been published by Cochrane, W.P.(Chemical derivatisation in pesticide analysis, Plenum Press, NY (1981)). Chemical reactions have the advantages of being fast and easy to carry out, but specialised reagents may need to be purchased and/or purified.
(b) Physical reactionsA useful technique is the photochemical alteration of a pesticide residue to give one or more products with a reproducible chromatographic pattern. A sample of standard pesticide and fortified extract should always be treated in a similar manner. Samples containing more than one pesticide residue may give problems in the interpretation of results. In such cases pre-separation of specific residues may be carried out using TLC, HPLC or column fractionation prior to reaction.
(c) Other methodsMany pesticides are susceptible to degradation/transformation by enzymes. In contrast to normal chemical reactions, these processes are very specific and generally consist of oxidation, hydrolysis or de-alkylation. The conversion products possess different chromatographic characteristics from the parent pesticide and may be used for confirmatory purposes if compared with reaction products using standard pesticides.
4.9 The concept of Lowest Calibrated Levels (LCL)
4.9.1 When the objective of the analysis is to monitor and verify the compliance with MRLs or other accepted limits (AL), the residue methods must be sufficiently sensitive to reliably determine the residues likely to be present in a crop or an environmental sample at or around the MRL or AL. However, for this purpose it is not necessary to use methods with sufficient sensitivity to determine residues at levels two or more orders of magnitude lower. Methods developed to measure residues at very low levels usually become very expensive and difficult to apply. The use of LCL (see Glossary) would have the advantage of reducing the technical difficulty of obtaining the data and would also reduce costs. The following proposals for LCLs in various samples may be useful in enabling the residue chemist to devise suitable methods.
4.9.2 For registered active ingredients with agreed MRLs, the LCL can be specified as a fraction of the MRL. For analytical convenience this fraction will vary and could be as follows:
| MRL
(mg/kg) | LCL
(mg/kg) |
|
5 or greater | 0.5 |
| 0.5 up to 5 |
0.1 increasing to 0.5 for higher MRLs |
|
0.05 up to 0.5 | 0.02 increasing
to 0.1 for MRLs |
|
less than 0.05 | 0.5 x MRL |
4.10 Expression of results
For regulatory purposes, only confirmed data should be reported, expressed as defined by the MRL. Null values should be reported as being less than lowest calibrated level, rather than less than a level calculated by extrapolation. Generally results are not corrected for recovery, and they may only be corrected if the recovery is significantly different from 100%. If results are reported corrected for recovery, then both measured and corrected values should be given. The basis for correction should also be reported. Where positive results obtained by replicate determinations (e.g. on different GC columns, with different detectors or based on different ions of mass spectra) of a single test portion (sub-sample), the lowest valid value obtained should be reported. Where positive results derive from analysis of multiple test portions, the arithmetic mean of the lowest valid values obtained from each test portion should be reported. Taking into account, in general, a 20-30% relative precision, the results should be expressed only with 2 significant figures (e.g.: 0.11, 1.1, 11 and 1.1 x 102). Since at lower concentrations the precision may be in the range of 50%, the residue values below 0.1 should be expressed with one significant figure only.
Figure II.1. Overview of Method Validation
Figure II.2. Verification of Analyte Stability
Table 1 Summary of parameters to be assessed for method validation
|
Parameters to be tested |
Existing analytical method, for which previous tests of the
parameter have shown that it is valid for one or more analyte/matrix combinations |
Modification of an existing method |
New method, not yet validated |
Experiment types which may be combined | ||||
| Performance verification* |
Additional matrix |
Additional analyte |
Much lower concentration of analyte |
Another laboratory | ||||
|
Specificity (show that the detected signal is due to the analyte, not another
compound) | No (provided criteria
for matrix blanks and confirmation of analyte are met) |
Yes, if interference from matrix is apparent in QC |
Yes | Yes, if interference from
matrix is apparent in QC | Rigorous
checks not necessary if the performance of the determination system is similar
or better | Yes or No. Rigorous
checks may be necessary if the determination system is fundamentally different
or where the extent of interferences from the matrix is uncertain |
Yes. Rigorous checks may be necessary if the determination system is different
or where the extent of interferences from the matrices are uncertain, compared
with existing methods | |
| Analytical Range, Recovery
through extraction, clean-up, derivatisation and measurement |
Yes | Yes |
Yes | Yes |
Yes | Yes |
Yes | Calibration range |
|
Calibration range for determination of analyte |
No | No |
Yes | Yes |
Yes, for representative analytes |
Yes, for representative analytes |
Yes, for representative analytes |
Linearity, reproducibility and signal/noise |
| LOD and LOQ |
No | Yes, (partial if matrix
is from a represented class) | Yes,
partial for represented analytes |
Yes | Yes |
Yes | Yes |
Lowest calibrated level, and low level spike recovery data |
| Reporting Limit, LCL |
Yes | No |
No | No |
No | No |
No | |
| Analyte stability in sample extracts*¡ |
No | Yes, unless matrix is from
a represented class | Yes, unless
the analyte is represented | Yes |
No | No, unless extraction/final
solvent is different, or the clean-up is less stringent |
Yes, if extraction/final solvent is different from that used in an existing
method, or the clean-up is less stringent, compared with existing methods used. |
|
| Analyte
stability dur- ing sample storage*Ä |
Yes | Yes |
Yes, | Ideally |
No | No |
No | |
| Extraction efficiency*¨ |
No | Ideally |
Ideally | Ideally |
No | No, unless different extraction
conditions employed | Yes, unless
previously tested extraction procedure is used. |
|
| Homogeneity*
of analytical samples | Yes§ |
No, unless the matrix is substantially different |
No | No |
No, unless the equipment is changed |
No, unless the equipment is changed |
Yes, unless a previously tested sample processing procedure is used |
See below |
|
Analyte stability in sample processing* |
No | Yes, unless a represented
matrix | Yes, unless a represented
analyte | Ideally |
No | No, unless procedure
involves higher temperature, longer time, coarser comminution, etc. |
No, unless procedure involves higher temperature, longer time, finer comminution,
etc. | Repeatability, reproducibility |
| than validated procedures. | ||||||||
* On-going quality controlTable 2 Parameters to be assessed for method validation in various circumstances* If relevant information is not available
¡ Representative analytes may be chosen on the basis of hydrolysis, oxidation and photolysis characteristics
Ä Stability data in/on representative commodities should provide sufficient information. Additional tests are required, for example, where: a samples are stored beyond the time period tested (eg. stability tested up to 4 weeks and measurable analyte loss occurs during this period, samples not analyzed until 6 weeks),
b stability tests were performed at £ - 18° C, but the samples are stored in the laboratory at £ 5° C;
c samples are normally stored at £ - 15° C, but storage temperature rises to + 5° C.
¨ Information on efficiency of extraction may be available from the manufacturer or company that is registering the compound.
§ Occasionally with repeated analysis of test portions of positive samples.
| Parameter |
Level(s) | No. of analyses or type of test required | Quantitative method | Criteria |
Comments | ||
|
Screening method | |||||||
|
1. Within-Laboratory (single laboratory) performance of optimised method | |||||||
| 1.1 Analyte stability
in extracts and standard solutions |
At £ AL, or with well detectable residues |
³ 5 replicates at each appropriate point
in time (including zero) and for each representative analyte/commodity. Fortify
blank sample extracts to test stability of residues. Compare analyte concentration
in stored and freshly made standard solutions. |
No significant change in analyte concentration in stored extracts and analytical
standards (P = 0.05) | At the end
of the storage period, residues added at LCL are detectable |
The test of stability in extracts is required if the analytical method is suspended
during the determination process, and the material will likely be stored longer
than during determination of precision, or if low recoveries were obtained during
optimisation of the method. During method optimisation, recovery should be measured
against both “old” and “freshly prepared” calibration standards,
if the recovery extracts are stored. Storage time should encompass the longest
period likely to be required to complete the analysis. | ||
| 1.2 Calibration function Matrix effect |
LCL to 2 (3) times AL | Test
the response functions of all analytes included in the method with ³
2 replicates at ³ 3 analyte levels plus blank sample. For non-linear
response, determine response curve at ³ 7
levels and ³ 3 replicates. Test the matrix
effect with all representative analytes and matrices. Apply the standards prepared
in solvent and sample extracts randomly. |
For linear calibration: regression coefficient for analytical standard solutions
® ³ 0.99. he SD of residuals (Sy/x)
£ 0.1 | For
linear calibration: regression coefficient ® ³ 0.98. SD of residuals £ 0.2 |
Calibration parameters may be established during optimisation of the procedure,
determination of precision or detection capability. Prepare calibration solutions
of different concentrations | ||
|
1.3 Analytical range, accuracy, trueness precision, limit of detection (LD),
limit of quantitation (LOQ) | LCL
to 2 (3) times AL* | Analyse representative
analyte matrix combinations: ³ 5 analytical portions spiked at zero, LCL, AL and
³ 3 replicates at 2-3 AL level. The recovery
tests should be divided among the analysts, who will use the method, and instruments
that will be involved in the analysis. |
LOQ should be fit for purpose. Mean recovery and CVA see Table 2. | All
recoveries are detectable at LCL |
The analysts should demonstrate that the method is suitable for determining
the presence of the analyte at the appropriate AL with the maximum (false negative
and false positive) errors specified. | ||
| 1.4 Specificity and selectivity of analyte
detection | At lowest calibration
level (LCL) | Identify by mass
spectrometry, by a similarly specific technique, or by the appropriate combination
of separation and detection techniques available. Analyse ³
5 blanks of each representative commodity obtained preferably from different
sources, Report analyte equivalent of blank response. |
Measured response is solely due to the analyte. Residues measured on two different
columns should be within the critical range of replicate chromatographic determinations. |
The rate of false negative samples (b error) at AL should typically be < 5%.. |
Applies only to a specific combination of separation and detection technique.
Samples of known treatment history may be used instead of untreated samples, for
analytes other than that applied during treatment. Maturity of sample matrices
may significantly affect the blank sample response. Blank values shall also be
regularly checked during performance verification (see Section 5 below). | ||
|
1.5 Selectivity of separation |
At AL | Determine RRt values
for all analytes to be tested by the method (not only the reference compounds).
When chromatographic techniques are used without spectrometric detection, apply
different separation principles and/or determine RRt-s on columns of different
polarity. Determine and report resolution (RS) and tailing factors
(Tf) of critical peaks. |
The nearest peak maximum should be separated from the designated analyte peak
by at least one full width at 10% of the peak height, or more selective detection
of all analytes is required. | Tentative
identification of all analytes tested. (Not all analytes need to be separated) |
Unless the chromatographic separation and spectrometric detection is used in
combination, report RRt values on columns of different polarity, which enable
the separation (minimum R ³ 1.2) of all analytes tested. | ||
| 1.6
Homogeneity of analyte in analytical sample |
At about AL or well detectable residues |
Analyse ³ 5 replicate test sample portions
of one representative commodity from each group (Table 4), post- processing. Determine
CVSp with analysis of variance. The analyte homogeneity should be checked
with analytes known to be stable. |
CVSp £ 10%. |
CVSp £ 15% |
Use preferably commodities with incurred stable surface residues or
treat the surface of a small portion of the natural units (<20%) of laboratory
sample before cutting or chopping to represent worst scenario of sample processing.
Processing validated for use with any subsequent procedure. Validation applicable
to other commodities with similar physical properties, and it is independent of
the analyte. The test may be combined with testing stability of analyte (see Section
1.7 of this Table) | ||
| 1.7 Analyte stability
during sample processing | About
AL | Fortify commodities with known
amounts of analytes before processing the sample. Analyse ³
5 replicates of each commodity, post-processing, Apply a notionally stable
marker compound together with the analytes tested. For MRM and group specific
methods, GSM, several analytes, which can be well separated, can be tested together. |
The stability of the analyte need not be specified if the average overall recovery
of analyte added before sample processing (including procedural recovery) and
CVA are within the ranges specified in Table 2 Quantify stability if
the overall recovery and the procedural recovery is significantly different (P=0.05). |
Analyte added at LCL remains detectable after processing |
The temperature of the sample during processing may be critical. Processing
validated for use with any subsequent procedure. Validation may be specific to
analyte and/or sample matrix. | ||
| 1.8 Extraction efficiency |
About AL or readily measurable residues |
Analyse ³ 5 replicate portions of samples
or reference material with incurred residues. |
For samples with incurred residues, the mean result obtained with the reference
procedure and the tested procedure should not differ significantly at P=0.05 level
applying CVL in the calculation. Or, the consensus value of reference
material and the mean residue should not differ significantly at P=0.05 level
when calculated with CVA of the method tested. When the CVA
of the method is larger than 10%, the number of replicate analyses has to be increased
to keep the relative standard error of the mean < 5%. |
The mean incurred residues, known to be present at or about the LOQ or LCL,
are actually detectable in the samples. |
Temperature of the extract, speed of blender or Ultra Turrax, time of extraction
and solvent/water/matrix ratio may significantly effect the efficiency of extraction.
The effect of these parameters can be checked with ruggedness test. The optimised
conditions should be kept constant as far as possible. | ||
| 1.9 Analyte stability
during sample storage | About AL |
Analyse freshly homogenised samples containing incurred residues, or homogenise
and spike blank samples (time 0), and then analyse samples stored according to
normal procedures of the laboratory (usually at £
-18° C). The storage time should be ³
than the longest interval foreseen between sampling and analysis. ³
5 replicates at each time point. When the stored portions are analysed ³ 4 occasions, test ³ 2 spiked portions, and ³ 1 blank portion spiked at the time of analysis. |
No significant loss of analyte during storage (P = 0.05) |
Analyte added at lowest calibration level, LCL, remains detectable after storage |
Storage is validated for use with any subsequent procedure. Validation is specific
to analyte. However, generally storage stability data obtained with representative
sample matrices can be considered valid for similar matrices. The matrices shall
be selected taking into account the chemical stability (e.g. hydrolysis) of the
analyte and the intended use of the substance. Useful information can be obtained
on stability during storage from the JMPR evaluations[32]
or from dossiers submitted for registration Report the initial residue concentration,
the remaining residue concentration and the procedural recovery of the analyte.
Unnecessary sample storage can be avoid by a careful planning for sampling and
consequent analysis through administrative arrangement, which is not a part of
analytical method. | ||
|
2. Extension of the validated method | |||||||
| 2.1 Analyte stability during sample storage,
processing, and in extracts and standard solutions. |
See 1.1, 1.2 & 1.9 | |
| |
Only if information on stability under the processing conditions and on the
representative matix is not already available | ||
| 2.2 Calibration function, matrix effect |
LCL to 2 (3) AL: | Three point
calibration embracing AL with and without matrix matched analytical standards |
For linear calibration: regression coefficient for analytical standard solutions
® ³ 0.99. SD of relative residuals (Sy/x)
£ 0.1 |
For linear calibration: regression coefficient ® ³ 0.98. SD of relative residuals £ 0.2 |
The method validation may not give definite information for the matrix effect,
because matrix effects change with time, with sample (sometimes), with column,
etc. | ||
| 2.3
Accuracy, precision, LOD, LOQ | at
AL | Planned in advance: |
The residues recovered should be within the repeatability limits of the method: |
Analytes added to blank samples at target reporting level should be measurable
in all tests. | Use CVAtyp
established during method validation. | ||
| 2.4 Specificity and
selectivity of analyte detection |
At LCL | Identify by mass spectrometry,
or by the appropriate combination of separation and detection techniques available. |
Measured response is solely due to the analyte. The detection system used should
have equal or better detector performance than those applied during method validation. |
The rate of false negative samples (b error) at AL should be < 5%.. |
When the extension for a new analyte is planned, the applicability of the method
shall be checked for all representative sample matrices in which the analyte may
occur. When an analyte is unexpectedly detected, the performance check may be
carried out for the actual matrix alone See also 1.4. | ||
|
2.5 Selectivity of separation |
See 1.5 | See 1.5 |
See 1.5 | See 1.5 |
See 1.5 Only if information is not available | ||
| 2.6 Extraction Efficiency |
See 1.8 | See 1.8 |
See 1.8 | See 1.8 |
See 1.8 Only if information is not available | ||
| 3. Adaptation of the validated method
in another laboratory | |||||||
|
3.1 Purity and suitability of chemicals, reagents and ad(ab)sorbents |
| Test reagent blank, applicability
of ad(ab)sorbents and reagents. Perform derivatization without and with sample. |
No interfering response |
No interfering response |
Some of the most common problems inmethod transfer involve differences inselection
of reagents, solvents andchromatographic media, or in equipment capabilities.
Whenever possible, try to confirm actual materials and equipment used by the method
developer, if that information is not provided with the method or publication,
as received. Substitutions can be tried after the method is working within your
laboratory. | ||
|
3.2 Analyte stability in extracts and standard solutions |
See 1.10 | See 1.1 |
See 1.1 | See 1.1 |
This testing may be omitted if full information on analyte stability is provided
with the method or if the method is replacing a previously used method for the
analyte and the stability information has been previously generated for the previous
method. | ||
|
3.3 Calibration function Matrix effect |
LCL to 2 (3) times AL | Test
the response functions of representative analytes included in the method at ³
3 analyte levels plus blank. For non-linear response, determine response curve
at ³ 7 levels and ³
3 replicates. |
For linear calibration: regression coefficient for analytical standard solutions
® ³ 0.99. The SD of relative residuals(Sy/x)
£ 0.1 |
For linear calibration: regression coefficient ® ³ 0.98. The SD of relative residuals £ 0.2 |
Sees: 1.2 | ||
|
3.4 Analytical range Accuracy and precision, limit of detection, limit of quantitation |
Blank extract and or AL | Analyse
representative analyte/matrix combinations: ³ 5 analytical portions each of blank samples spiked
at 0 and AL, and 3 portions spiked at 2 AL. | Average recovery
and CVA should be within the ranges given in Table 2. |
All recoveries detectable at LCL. |
See comments in 1.3. | ||
|
3.5 Specificity and selectivity of analyte detection |
At AL | Check performance characteristics
of detectors used and compare them with those specified in the method. Check response
of one blank of each representative commodity, otherwise perform test as described
in section 1.4. | Measured response
is solely due to the analyte. |
The rate of false negative samples (b error) at AL should typically be < 5%. |
The relative response of specific detectors can substantially vary from model
to model. Proper checking of specificity of detection is critical for obtaining
reliable results. Compare blank response observed with typical peaks reported
in blank extracts See other comments under section 1.4. | ||
| 3.6 Analyte“homogeneity” |
At about AL or well detectable residues |
Test two representative commodities of different nature |
CVSp<10% | CVSp<15% | The tests are
performed to confirm similarity of application conditions and applicability of
parameters obtained by the laboratory validating the method. When the test results
in similar CV Sp as reported, the conditions of sample processing
may be considered similar and further tests are not required for the validation
of the method. | ||
|
3.7 Analyte stability in extracts and standard solutions |
See 1.1 | See 1.1 |
See 1.1 | See 1.1 |
This testing may be omitted if full information on analyte stability is provided
with the method or if the method is replacing a previously used method for the
analyte and the stability information has been previously generated for the previous
method. | ||
| Concentration |
Repeatability | Reproducibility |
Trueness[34] | ||
| CVA%[35] |
CVL%[36] |
CVA%[37] |
CVL%[38] |
Range of mean % recovery | |
| £
1 mg/kg | 35 |
36 | 53 |
54 | 50-120 |
| >
1 mg/kg £ 0.01 mg/kg |
30 | 32 |
45 | 46 |
60-120 |
|
> 0.01 mg/kg £ 0.1 mg/kg |
20 | 22 |
32 | 34 |
70-120 |
|
> 0.1 mg/kg £ 1 mg/kg |
15 | 18 |
23 | 25 |
70-110 |
|
> 1 mg/kg | 10 |
14 | 16 |
19 | 70-110 |
| Parameter |
Level (s) | No. of analyses or type of test required | Quantitative method | Criteria |
Comments |
|
Screening method | |||||
|
4. Quality control (performance verification) | |||||
| 4.1 Methods used regularly | |||||
| 4.1.1 Suitability of
chemicals, adsorbents and reagents |
| For each new batch: Test
reagent blank, applicability of ad(ab)sorbents and reagents Perform derivatization
without sample. | No interfering
response³ 0.3 LCL. |
No interfering response ³ 0.5AL. |
Alternately, if the sample blank, calibration and the recovery are satisfactory
then the suitability of reagents etc are confirmed. |
| 4.1.2 Calibration and analytical range |
| Single point calibration
may be used with standard mixtures, if the intercept of calibration function is
close to 0. Apply multi point calibration (3 x 2) for quantitative confirmation. |
The analytical batch may be considered to be under statistical control if the
analytical standards and sample extracts are injected alternately, and the calculated
SD of relative residuals is £ 0.1. |
Analyte is detected at LCL. | Standard
solution and samples should be injected alternately. |
| 4.1.3 Accuracy and
precision | Within analytical range |
Include in each analytical batch ³ 1 sample either: |
The performance of detector and chromatographic column shall be equal or better
than specified in the method. | Fortify analytical
portion with standard mixture(s). Alter standard mixtures in different batches
to obtain recoveries for all analytes of interest at regular intervals. Perform
alternately recovery studies at AL as well as at LCL and 2 times AL, as appropriate,
to confirm applicability of the method within the analytical range. The frequency
of recovery studies at AL should be 2 to 3 times higher then those at other levels. | |
| 4.1.4 Selectivity of separation, Specificity
of detection Performance of detectors |
| Include appropriate detectiontest
mixture in each chromatography batch. Include untreated commodity (if available)
in analytical batch. Use standard addition if no untreated sample (similar to
those analysed in the batch) is available |
Rs, Tf of test compounds, and RRF and d
of the detection should be within the Specified range. RRt-s should be within
2% for GLC and 5 % for HPLC determinations Detector performance should be within
specified range. Sample co-extractives interfering with the analyte should not
be present ³ 0.3 LCL. The recovery of added standard should be
within the acceptable recovery range of the analyte. |
Detector performance should be within specified range. Analyte should be seen
above LCL or CCa for banned compounds. |
This is also sometimes referred to as a“system suitability” test.
Prepare detection test mixture for each method of detection. Select the components
of the mixture in order to indicate the characteristic parameters of chromatographic
separation and detection. |
| 4.1.5 Analyte homogeneity
in processed sample | At well detectable
analyte concentration. | Select
a positive sample randomly. Repeat analysis of another one or two analytical portions. |
The residues measured on two different days should be within the reproducibility
limit of replicate analytical portions: |
Perform test alternately to cover each commodity analysed. Test homogeneity
at the beginning of growing season, or at the start of the analysis of the given
type of samples. | |
| 4.1.6 Extraction efficiency |
| |
| |
The efficiency of the extraction cannot be controlled during the analysis.
To ensure appropriate efficiency, the validated extraction procedure should be
carried out without any change. |
|
4.1.7 Duration of analysis | |
| The samples, extracts etc.
should not be stored longer than the period for which the storage stability was
tested during method validation. Storage conditions should be regularly monitored
and recorded. | Examples for the
need of additional storage stability tests are given under Table 2. | |
| 4.2 Analyte detected
occasionally | |||||
|
FOLLOW TESTS DESCRIBED IN 4.1 WITH THE FOLLOWING EXCEPTIONS | |||||
| 4.2.1 Accuracy and precision |
At around AL | Reanalyse another
analytical portion; |
The residues measured on two different days should be within the critical range: | Check accuracy
if residue found at ³ 0.5AL. | |
|
4.3 Methods used at irregular intervals | |||||
| Follow tests described in 4.1 with the
following exceptions | |||||
|
4..3.1 Accuracy and precision (repeatability) |
At AL and LCL | Include one
fortified sample at LCL and two samples at AL in each analytical batch. Use standard
addition if untreated sample (similar to those analysed in the batch) is not available. |
Minimum two recoveries shall be within warning limit, one may be within action
limit. |
The acceptable results also prove the suitability of chemicals, adsorbents
and reagents used. | |
|
4.4. Changes in implementation of the method | |||||
| Change |
Parameters to be tested | For
test methods and acceptability criteria see the appropriate sections of Appendix
1. | |||
| 4.4.1
Chromatogra phic column | Test
selectivity of separation, resolution, inertness, RRt values |
Performance characteristics should not be affected |
Apply appropriate test mixtures to obtain information on the performance of
the column. | ||
|
4.4.2 Equipment for sample processing |
Homogeneity of processed sample; |
Test described in 1.6 and 1.7 shall be performed and they should give results
conforming with the relevant criteria.. |
Homogeneity test is only necessary if the degree of comminution and/or mixing
is inferior to that of the original equipment. The stability of analytes need
to be tested if the processing time and temperature are significantly increased. | ||
| 4.4.3 Equipment for
extraction | Compare field incurred
residue levels detected with the old and new equipment in ³
5 replicates | The mean residues
should not be significantly different at p = 0.05 level. |
Test is necessary if a new type of equipment is used | ||
| 4.4.4 Detection |
Test selectivity of separation and selectivity and sensitivity of detection |
Performance characteristics should be the same or better specified in the description
of the method. | Test also detectability
separately with new detection reagents. | ||
|
4.4.5 Analyst | ³
5 recovery tests at each level (LCL, AL and 2 (3) AL), re-analysis of one
blank sample and two positive samples (unknown to the analyst) |
All results should be within the warning limits specified for the method in
the laboratory. Replicate sample analysis shall be within the critical range. |
This is a minimum requirement. Laboratories in some areas of residue work use
a more detailed protocol which includes: (1) generation of standard curve within
acceptability criteria; (2) minimum of 2 analytical runs for each matrix, containing
representative analytes fortified by the analyst at a minimum of 3 levels in duplicate;
(3) minimum of 1 analytical run containing fortified or incurred samples, 3 levels
in duplicate, provided as unknowns to the analyst. All results must meet acceptability
criteria, or be repeated. | ||
|
4.4.6 Laboratory | Accuracy
and precision ³ 3 recovery tests at each
level (LCL, AL and 2 (3) AL) by (different) analyst(s) on different days. |
All results should be within the warning limits specified for the method in
the laboratory. | The reproducibility
of the method under the new conditions must be established and it has to be done
by more than one analyst if available. | ||
| Commodity
Group | Common properties |
Commodity class[39] |
Representative species |
| Plant products | |||
| I. |
High water and chlorophyll content |
Leafy vegetables Brassica |
spinach or lettuce |
| II. |
High water and low or no chlorophyll content |
Pome fruits, | apple, pear |
|
III. | High acid content |
Citrus fruits | orange, lemon |
| IV. |
High sugar content | |
raisins, dates |
|
V. | High oil or fat |
Oil seeds | avocado,
sunflower seed |
| VI. |
Dry materials | Cereals |
wheat, rice or maize grains |
|
Cereal products | wheat bran,
wheat floor | ||
|
Commodities requiring individual test |
| e.g. garlic, hops, tea, spices,
cranberry | |
|
Products of animal origin | |||
|
|
|
Meats | Cattle meat, chicken
meat |
| Edible
offals | Liver, kidney | ||
| Fat |
Fat of meat | ||
|
Milk | Cow milk | ||
| Eggs |
Chicken egg | ||
Note: The method should be validated with representative pesticides for each commodity group. Commodities which are difficult to analyse require individual tests.Table 6. Examples of detection methods suitable for the confirmatory analysis of substances
| Detection
method | Criterion |
| LC or GC and Mass spectrometry |
if sufficient number of diagnostic ions are monitored |
| LC-DAD or scanning UV |
if the UV spectrum is characteristic |
|
LC - fluorescence | in combination
with other techniques |
|
2-D TLC - (spectrophotometry) |
in combination with other techniques |
|
GC-ECD, NPD, FPD | only if combined
with two or more separation techniques[40] |
| Derivatisation |
if it was not the first choice method |
|
LC-immunogram | in combination
with other techniques |
|
LC-UV/VIS (single wavelength) |
in combination with other techniques |
Glossary of terms
|
Accepted Limit (AL) | Concentration
value for an analyte corresponding to a regulatory limit or guideline value which
forms the purpose for the analysis, e.g. MRL, MPL; trading standard, target concentration
limit (dietary exposure assessment), acceptance level (environment) etc. For a
substance without an MRL or for a banned substance there may be no AL (effectively
it may be zero or there may be no limit) or it may be the target concentration
above which detected residues should be confirmed (action limit or administrative
limit). |
|
Accuracy | Closeness of agreement
between a test result and the accepted reference value. |
| Alpha (a)
Error | Probability that the true
concentration of analyte in the laboratory sample is less than a particular value
(e.g. the AL) when measurements made on one or more analytical/test portions indicate
that the concentration exceeds that value (false positive). Accepted values for
this probability are usually in the range 1 to 5%. |
| Analyte |
The chemical substance sought or determined in a sample. |
| Analyte Homogeneity (in sample) |
Uniformity of dispersion of the analyte in matrix. The variability in analyticalresults
arising from sample processing depends on the size of analytical portion. The
sampling constant[41] describes the relationship
between analytical portion sizeand the expected variation in a well mixed analytical
sample: KS = w (CVSp)[42],
where w is the mass of analytical portion and CVSp is the coefficient
of variation of the analyte concentration in replicate analytical portions of
w (g)which are withdrawn from the analytical sample |
| ANALYTICAL PORTION |
A representative quantity of material removed from the analytical sample, of
proper size for measurement of the residue concentration. |
| ANALYTICAL SAMPLE |
The material prepared for analysis from the laboratory sample, by separation
of the portion of the product to be analysed and then by mixing, grinding, fine
chopping, etc., for the removal of analytical portions with minimal sampling error. |
| APPLICABILITY |
The analytes, matrices and concentrations for which a method of analysis has
been shown to be satisfactory. |
|
Beta (b) Error |
Probability that the true concentration of analyte in the laboratory sample
is greater than a particular value (e.g. the AL) when measurements made on one
or more analytical portions indicate that the concentration does not exceed that
value (false negative). Accepted values for this probability are usually in the
range 1 to 5%. |
|
Bias | Difference between the
mean value measured for an analyte and an accepted reference value for the sample.
Bias is the total systematic error as contrasted to random error. There may be
one or more systematic error components contributing to the bias. A larger systematic
difference from the accepted reference value is reflected by a larger bias value. |
| Commodity Group |
Group of foods or animal feeds sharing sufficient chemical characteristics
as to make them similar for the purposes of analysis by a method. The characteristics
may be based on major constituents (e.g. water, fat, sugar, and acid content)
or biological relationships, and may be defined by regulations. |
| Confirmatory Method |
Methods that provide complete or complementary information enabling the analyte
to be identified with an acceptable degree of certainty [at the Accepted Limit
or level of interest]. As far as possible, confirmatory methods provide information
on the chemical character of the analyte, preferably using spectrometric techniques.
If a single technique lacks sufficient specificity, then confirmation may be achieved
by additional procedures consisting of suitable combinations of clean-up, chromatographic
separation(s) and selective detection. Bioassays can also provide some confirmatory
data. |
| Decision Limit (CCa) |
Limit at which it can be decided that the concentration of the analyte present
in a sample truly exceeds that limit with an error probability of a
(false positive). In the case of substances with zero AL, the CCa
is the lowest concentration level, at which a method can discriminate with a statistical
probability of 1 - a whether the identified analyte
is present. The CCa is equivalent to the limit of
detection (LOD) under some definitions (usually for a
= 1%). |
| Detection Capability (CCb) | Smallest
true concentration of the analyte that may be detected, identified andquantified
in a sample with a beta error (false negative). In the case of banned substances
the CCb is the lowest concentration at which a method
is able to determine the analyte in contaminated samples with a statistical probability
of |
|
Detection Test Mixture | Mixture
of analytical standards which are suitable to check the conditions of chromatographic
separation and detection. The detection test mixture should contain analytes which
provide information for the selectivity and response factors for the detectors,
and the inertness (e.g. characterised by the tailing factor Tf) and separation
power (e.g. resolution Rs) of column, and the reproducibility of RRt. The detection
test mixture may have to be column and detector specific. |
| False negative result |
See beta error |
|
False positive result | See
alpha error |
|
Incurred Residue | Residues
of an analyte in a matrix arising by the route through which the trace levels
would normally be expected, as opposed to residues from laboratory fortification
of samples. Also weathered residue. |
|
Individual Method | Method which
is suitable for determination of one or more specified compounds. A separate individual
method may be needed, for instance to determine some metabolite included in the
residue definition of an individual pesticide or veterinary drug. |
| Laboratory Sample |
The sample as received at the laboratory (not including the packaging). |
| Limit of Detection
(LD) | Smallest concentration where
the analyte can be identified. Commonly defined as the minimum concentration of
analyte in the test sample that can be measured with a stated probability that
the analyte is present at a concentration above that in the blank sample. IUPAC
and ISO have recommended the abbreviation LD. See also Decision Limit. |
| Limit of Quantitation
(LOQ) | Smallest concentration
of the analyte that can be quantified. Commonly defined as the minimum concentration
of analyte in the test sample that can be determined with acceptable precision
(repeatability) and accuracy under the stated conditions of the test. See also
Detection Capability. |
|
Lowest Calibrated Level (LCL) |
Lowest concentration of analyte detected and measured in calibration of the
detection system. It may be expressed as a solution concentration in the test
sample or as a mass and must not include the contribution from the blank |
| Matrix |
Material or component sampled for analytical studies, excluding the analyte. |
| Matrix Blank |
Sample material containing no detectable level of the analytes of interest. |
| Matrix-matched Calibration |
Calibration using standards prepared in an extract of the commodity analysed
(or of a representative commodity). The objective is to compensate for the effects
of co-extractives on the determination system. Such effects are often unpredictable,
but matrix-matching may be unnecessary where co-extractives prove to be of insignificant
effect. |
|
Method | The series of procedures
from receipt of a sample for analysis through to the production of the final result. |
| Method Validation |
Process of verifying that a method is fit for purpose. |
| Multi residue Method, MRM |
Method which is suitable for the identification and quantitation of a range
of analytes, usually in a number of different matrices. |
| Negative Result |
A result indicating that the analyte is not present at or above the lowest
calibrated level. (see also Limit of Detection) |
| Performance Verification |
Sets of quality control data generated during the analysis of batches of samples
to support the validity of on-going analyses. The data can be used to refine the
performance parameters of the method. |
|
Positive Result | A result indicating
the presence of the analyte with a concentration at or above the lowest calibrated
level. |
| Precision |
Closeness of agreement between independent test results obtained under stipulated
conditions. |
|
Quantitative Method | A method
capable of producing results, expressed as numerical values in appropriate units,
with accuracy and precision which fit for the purpose. The degree of precision
and trueness must comply with the criteria specified in Table 3. |
| Recovery |
Fraction or percentage of an analyte recovered following extraction and analysis
of a blank sample to which the analyte has been added at a known concentration
(spiked sample or reference material). |
|
Reagent Blank | Complete analysis
made without the inclusion of sample materials for QC purpose. |
| Reference Material |
Material one or more of whose analyte concentrations are sufficiently homogeneous
and well established to be used for the assessment of a measurement method, or
for assigning values to other materials. In the context of this document the term
"reference material" does not refer to materials used for the calibration of apparatus. |
| Reference Method |
Quantitative analytical method of proven reliability characterised by well
established trueness, specificity, precision and detection power. These methods
will generally have been collaboratively studied and are usually based on molecular
spectrometry. The reference method status is only valid if the method is implemented
under an appropriate QA regime. |
|
Reference Procedure | Procedure
of established efficiency. Where this is not available, a reference procedure
may be one that, in theory, should be highly efficient and is fundamentally different
from that under test. |
|
Repeatability | Precision under
repeatability conditions, i.e. conditions where independent test results are obtained
with the same method on replicate analytical portions in the same laboratory by
the same operator using the same equipment within short intervals of time. (ISO
3534-1) |
|
Representative Analyte | Analyte
chosen to represent a group of analytes which are likely to be similar in their
behaviour through a multi-residue analytical method, as judged by their physico-chemical
properties e.g. structure, water solubility, Kow, polarity,
volatility, hydrolytic stability, pKa etc. |
|
Represented Analyte | Analyte
having physico-chemical properties which are within the range of properties of
representative analytes. |
|
Reproducibility | Closeness
of agreement between results obtained with the same method on replicate analytical
portions with different operators and using different equipment (within laboratory
reproducibility). Similarly, when the tests are performed in different laboratories
the inter-laboratory reproducibility is obtained. |
| Representative Commodity |
Single food or feed used to represent a commodity group for method validation
purposes. A commodity may be considered representative on the basis of proximate
sample composition, such as water, fat/oil, acid, sugar and chlorophyll contents,
or biological similarities of tissues etc.. |
|
Ruggedness | Ability
of a chemical measurement process to resist changes in test results when subjected
to minor changes in environmental and method procedural variables, laboratories,
personnel, etc. |
|
Sample Preparation | The procedure
used, if required, to convert the laboratory sample into the analytical sample,
by removal of parts (soil, stones, bones, etc.) not to be included in the analysis. |
| Sample Processing |
The procedure(s) (e.g. cutting, grinding, mixing) used to make the analytical
sample acceptably homogeneous with respect to the analyte distribution, prior
to removal of the analytical portion. The processing element of preparation must
be designed to avoid inducing changes in the concentration of the analyte. |
| Screening Method |
A methods used to detect the presence of an analyte or class of analytes at
or above the minimum concentration of interest. It should be designed to avoid
false negative results at a specified probability level (generally b
= 5%). Qualitative positive results may be required to be confirmed by confirmatory
or reference methods. See Decision Limit and Detection Capability. |
| Selectivity |
Measure of the degree to which the analyte is likely to be distinguished from
other sample components, either by separation (e.g., chromatography) or by the
relative response of the detection system. |
|
Specificity | Extent
to which a method provides responses from the detection system which can be considered
exclusively characteristic of the analyte. |
|
Standard Addition | A procedure
in which known amounts analyte are added to aliquots of a sample extract containing
the analyte (its initially measured concentration being X), to produce new notional
concentrations (for example, 1.5X and 2X). The analyte responses produced by the
spiked aliquots and the original extract are measured and the analyte concentration
in the original extract (zero addition of analyte) is determined from the slope
and intercept of the response curve. Where the response curve obtained is not
linear, the value for X must be interpreted cautiously. |
| Tailing Factor |
Measure of chromatographic peak asymmetry; at 10% peak height maximum, the
ratio of the front and tail segments of peak width, when separated by a vertical
line drawn through the peak maximum. |
|
Test Portion | See “Analytical
Portion” |
|
Test Sample | See “Analytical
Sample” |
|
Trueness | Closeness of agreement
between the average value obtained from a large series of test results and an
accepted reference value. |
|
Uncertainty of measurement | Single
parameter (usually a standard deviation or confidence interval) expressing the
possible range of values around the measured result, within which the true value
is expected to be with a stated degree of probability. It should take into account
all recognised effects operating on the result, including: overall long-term precision
(within laboratory reproducibility) of the complete method; the method bias; sub-sampling
and calibration uncertainties; and any other known sources of variation in results. |
|
Cb |
See Annex IV |
MRL | Maximum
Residue Limit |
|
Cmax | See Annex
4 | MRM |
Multi-Residue Method |
|
Cmin | See Annex
4 | RRt |
Relative retention value for a peak |
|
CVAtyp | See Annex
4 | Rs |
Resolution of two chromatographic peaks |
|
CVLtyp | See Annex
4 | SD |
Standard Deviation |
|
CVSP | See Annex
4 | Sy/x |
Standard deviation of the residuals calculated from the linear calibration
function |
|
GLP | Good Laboratory Practice |
| |
| GSM |
Group Specific Method | |
|
| |
| WHO |
World Health Organization |
![]()
![]()
![]()
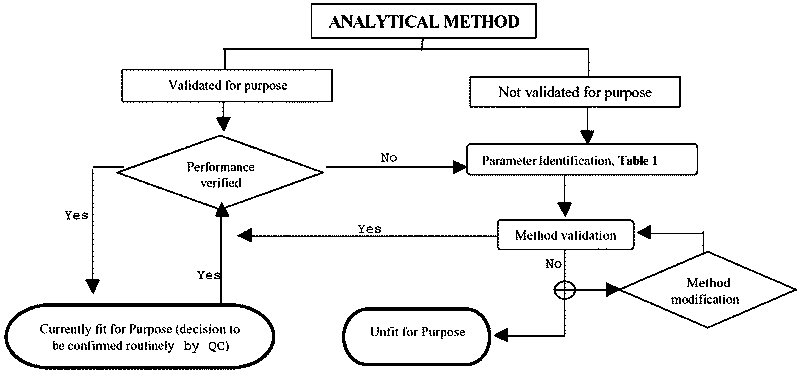{kind=link}
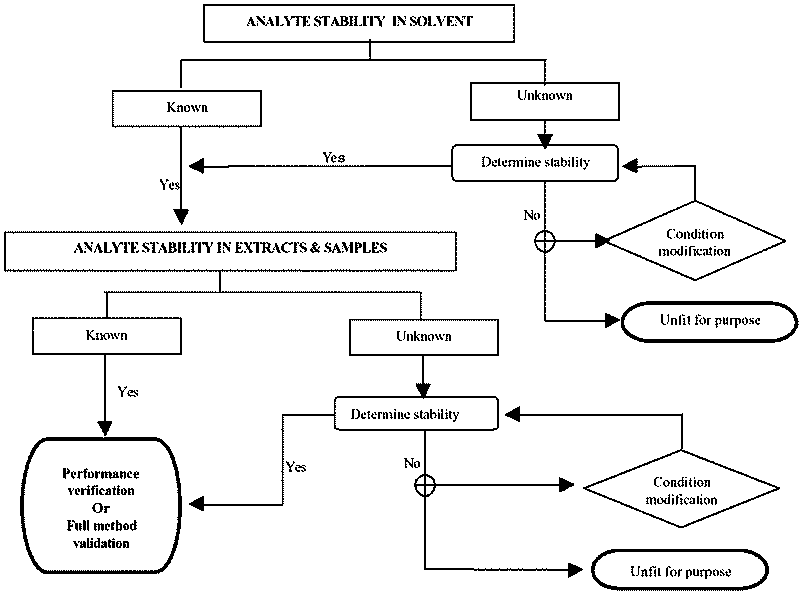{kind=link}