Isolation and analysis of double-stranded RNAs
D. Boscia
Double-stranded RNAs (dsRNAs) are paired molecules of viral genomic or subgenomic RNAs made up of a positive sense (+) RNA strand bound to a complementary negative sense (-) strand. dsRNAs represent replicative forms of viral RNAs which are formed during infection and may accumulate in the cells of diseased plants. They are stable molecule complexes with a size double that of genomic and subgenomic single-stranded RNAs and can be extracted from infected tissues. Their detection in plant extracts is regarded as a reliable indication of viral infection. By estimating the relative size of dsRNAs, it is possible to identify, with a fair approximation, the taxonomic group to which the eliciting virus belongs. However, specific identification of individual viruses is not possible. With grapevines, the study of dsRNA patterns is a useful complementary technique for determining the occurrence of non-mechanically transmissible, phloem-limited viruses.
Operations and materials for electrophoresis are the same as described for detection of viroids in the preceding chapter.
BUFFERS
PROCEDURE
1. Collect 10 to 15 g of cortical tissues from green or, preferably, mature canes and pulverize in a chilled mortar with liquid nitrogen.
2. Add 1 to 2 volumes of extraction buffer:
2X STE, 45 ml
10 percent SDS, 15 ml
Phenol (water saturated), 25 ml
NH4OH, 0.1 ml
Bentonite (40 mg per ml), 0.8 ml
2-Mercaptoethanol, 1 ml
3. Stir the mixture until amalgamation.
4. Add 25 ml of chloroform.
5. Stir at room temperature for 45 minutes.
6. Centrifuge at 8 000 rpm for 15 minutes.
7. Collect liquid phase by filtering through glass wool and make up to a volume of 100 ml.
8. Add 2 g of CF- 11 cellulose powder.
9. Add 22 ml of 95 percent ethanol to reach a final alcohol concentration of approximately 17 percent. Stir for 45 minutes at room temperature.
10. Put the suspension in a glass column (alternatively a 1 0-ml plastic disposable syringe is suitable), allow the cellulose powder to set, then wash with not less than 100 ml STE containing 16.5 percent ethanol.
11. Drain cellulose by pumping air gently with a piston from the top of the column. Remove the piston, making sure that the cellulose pad does not break. Elute dsRNA by flushing the cellulose pad three times with 3 ml of STE each time. (To secure maximum recovery it is advisable to drain the pad each time by pumping air with a piston as above.)
12. Centrifuge the eluted liquid at 8 000 rpm for 3 minutes to eliminate residual cellulose.
13. Add 20 ml of 95 percent cold ethanol and 0.5 ml of 3 M sodium acetate pH 5 and stir thoroughly.
14. Incubate overnight at -20°C or for 3 hours at 70°C
15. Centrifuge at 8 000 rpm for 30 minutes.
16. Save pellets and resuspend in 0.5 ml STE. Add 1 ml of cold 95 percent ethanol and 20 pi of 3 M sodium acetate pH 5, stir and leave standing at -70°C for 3 hours.
17. Centrifuge at 12 000 rpm for 15 minutes in an Eppendorf centrifuge, discard supernatant fluid and dry out pellets.
18. Resuspend pellets in 20 ?l of electrophoretic buffer and add 5 Ill of 0.3 percent bromophenol blue dissolved in 50 percent glycerol or sucrose solution.
19. Load on 6 percent polyacrylamide gel and apply a constant current of 30 mA for 6 hours or 20 mA overnight.
20. Remove gel from chamber and form, stain with ethidium bromide or silver nitrate and photograph.
REFERENCE
Fraenkel-Conrat, H., Singer, B. & Tsugita, A. 1961. Purification of viral RNA by means of bentonite. Virology' 14: 54-58.
D. Boscia and G.P. Martelli
Western blotting is an electrophoretic procedure whereby the coat protein subunits of phloem-limited, non-mechanically transmissible grapevine viruses can be separated from other components of plant extracts and identified following the reaction with specific antisera and staining. When molecular weight markers are used, the molecular weight of protein subunits can be determined and compared with known values. Specific recognition relies upon the use of homologous immunoglobulins.
Operations and materials for electrophoresis are the same as described previously for detection of viroids.
MATERIALS AND SOLUTIONS
PROCEDURE
1. Prepare polyacrylamide gels: 12 percent (separating gel) and 5 percent (stacking gel).
2. Put separating gel in the glass form, filling to about 2 cm from the top. Add distilled water gently and let stand for 30 minutes for polymerization.
3. Pour out distilled water, remove remaining water with filter paper, load stacking gel on top of polymerized separating gel and let stand for an additional 30 minutes for polymerization.
4. Connect power supply and make a blank electrophoretic run for 30 minutes at 30 V.
5. Prepare sample to be examined by grinding 2 to 5 g of cortical, petiolar or leaf vein tissues in a mortar with liquid nitrogen following the procedure outlined in the chapter on extraction of closteroviruses from grapevine tissues, below.
6. Resuspend high-speed pellets in 200 ml of 4X degradation buffer and heat at 100°C for 2 minutes.
7. Place samples in the wells and apply 130 V constant current for about 1 hour until the stain bands reach the bottom of the gel.
8. Wearing rubber gloves, remove the gel and immerse it for 30 minutes in transfer buffer. At the same time, soak polyvinylidene difluoride membrane (PVDF) (Immobilon P-Millipore) in 100 percent methanol and the filter paper in distilled water, then move both to transfer buffer.
9. When gel is ready, gently remove Trans Blot cover and steel cathode (semi-dry transfer cell, Bio Rad)(Figures 310 and 311).
10. Place filter paper on to plate-shaped anode and roll a glass rod gently on top of it to remove air bubbles (Figure 312).
11. Place gel slab on filter paper, making sure that there are no air bubbles.
12. Place PVDF membrane on the gel slab, making sure that the gel is centred and that there are no air bubbles. Cover gel with another layer of filter paper and roll a glass rod gently over it to remove air bubbles (Figures 313 to 315).
13. Carefully replace steel cathode and Trans Blot cover, and connect the apparatus to power supply (Figure 316).
14. Apply current as follows (Figure 317):
15. Remove PVDF membrane from Trans Blot and incubate for 1 hour at 37°C in PBS buffer containing 2 percent non-fat milk powder (2 g in 100 ml PBS buffer). Save 5 ml of this solution.
16. Place PVDF membrane in a polythene bag, and pipette into it 3 ml of PBS non-fat milk solution to which 3 ?g per ml of immunoglobulins have been added.
17. Remove air bubbles from plastic bag and seal. Incubate at 37°C for 2 hours or overnight at 4°C.
18. Wash PVDF membrane three times for 10 minutes in PBS-T and twice for 10 minutes in TBS-T.
19. Incubate PVDF membrane with a suspension of protein A-gold conjugate (gold enhancement kit, Bio Rad) at room temperature in the dark for 1 to 24 hours. The length of incubation depends on the intensity of a signal constituted by a faint pink band. If no signal appears after 24 hours proceed to the following steps.
20. Wash PVDF membrane twice for 10 minutes in TBS-T, twice for 10 minutes in TBS, five times for 1 minute in distilled water and once for 5 minutes in citrate buffer.
21. Prepare enhancement solution during the last washing.
22. Incubate PVDF membrane in enhancement solution at room temperature in the dark for 5 to 15 minutes. Rinse with distilled water.
23. Soak PVDF membrane in fixing solution for 5 minutes. Rinse with distilled water and dry with blotting paper. 24. Observe and record positions of dark-staining bands corresponding to dissociated viral coat protein subunits.
FIGURE 311 Disassembled Trans Blot apparatus showing plate-shaped electrodes
FIGURE 313 Gel slab is placed on filter paper
FIGURE 316 Steel cathode is in place
FIGURE 317 Trans Blot apparatus connected to power supply ready for operation
Detection of viruses and viroids by molecular
hybridization
Extraction of closteroviruses from grapevine
tissues
Extraction of phloem-limited isometric viruses
from grapevine tissues
Isolation and culture of Xylella fastidiosa
Detection of viruses and viroids by molecular hybridization
G. Macquaire, T. Candresse and J. Dunez
PRINCIPLE
A viral particle is composed of nucleic acids [ribonucleic acid (RNA) or deoxyribonucleic acid (DNA)] and a capsid made up of several dozen to a thousand copies of coat protein subunit. In some cases, the virus possesses an envelope composed of viral proteins integrated in membranes deriving from the host cell. Serological techniques detect the virus by specific recognition of the coat protein by specific antibodies developed in animals against this protein. Molecular hybridization techniques detect viral nucleic acids by specific recognition of their nucleotide sequence.
Nucleic acids are long, linear polymers of nucleotide molecules. Each nucleotide is in turn composed of several elements: a nitrogencontaining base linked to a phosphate group and a sugar molecule (ribose for RNA and deoxyribose for DNA). DNA contains four different bases: adenine (A), guanine (G), cytosine (C) and thymine (T). In the case of RNA, thymine is replaced by uracil (U), the three other bases being the same.
DNA is usually found in a double-stranded configuration, i.e. two chains of DNA associate through specific base pairing (A pairs with T and C pairs with G). Base pairing is extremely specific and creates non-covalent hydrogen bonds that unite the molecules associated in this way. RNA is most commonly found in a singlestranded configuration but, like DNA, it possesses the capacity to form double-stranded structures through A-U and G-C pairing.
The specific pairing of the bases composing nucleic acids constitutes the basis for the formation of hybrids (double-stranded structure) between complementary molecules and thus for the use of molecular hybridization as a diagnostic technique.
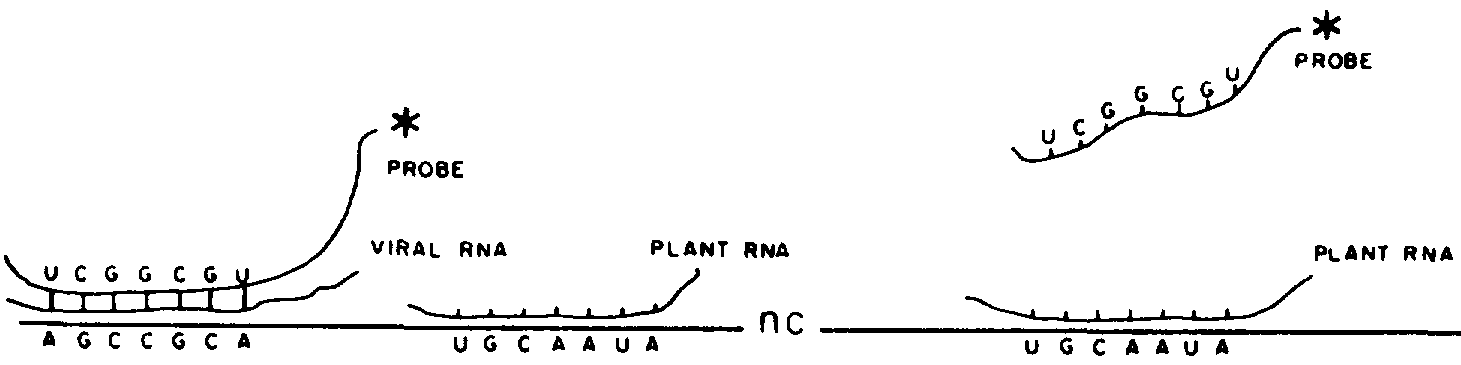
Nucleic acid molecules differ from one another in the order and sequence of alignment of their nucleotides (= nucleotide sequence). Two molecules of complementary sequences will form double-stranded hybrids under suitable conditions. Forexample, TCGGCGTAT will pair with AGCCGCATA to make a DNA-DNA hybrid.
A probe used for virus detection in molecular hybridization experiments is a single-stranded nucleic acid molecule prepared from a viral nucleic acid, with a nucleotide sequence complementary to that of the target viral RNA molecule.
Thus a DNA probe with the sequence TCGGCGTAT will specifically detect RNA and DNA molecules with the respective sequences AGCCGCAUA and AGCCGCATA. An RNA probe with the same specificity would be UCGGCGUAU.
The molecular hybridization detection system presented here is
based on a solid support hybridization, the samples being
permanently immobilized on a nitrocellulose membrane (Figure
318). We describe the technique using the two most frequently
used types of probe:
complementary DNA probes cloned in a plasmid vector;
in vitro transcribed complementary RNA probes prepared from
complementary DNA cloned into special purpose transcription
plasmid vectors.
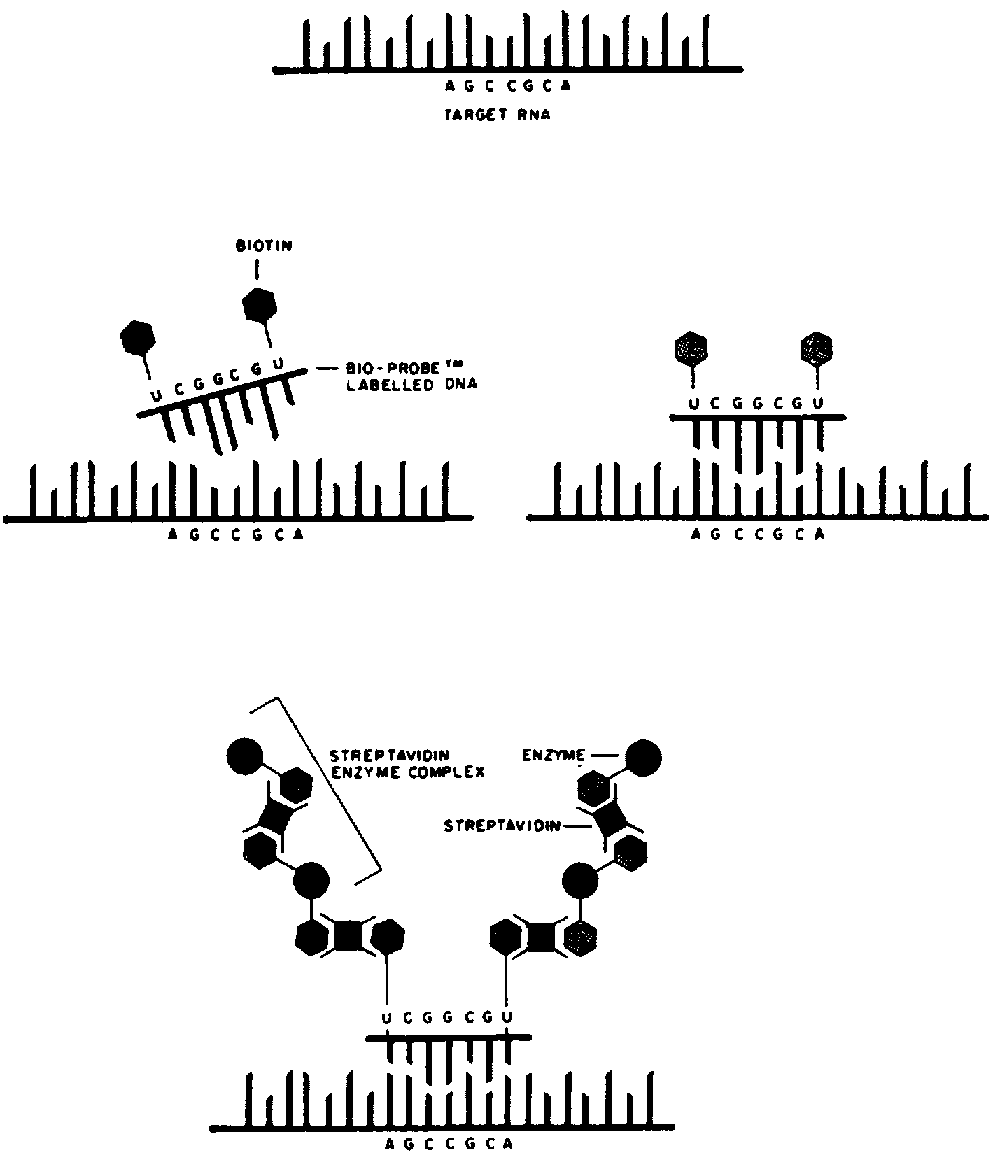
The probes can be labelled either radioactively or by incorporation of a nonradioactive marker such as biotin. The techniques for the determination of the probespecific activity are described following the experimental protocol.
BUFFERS
See Table 9 for commercial sources of chemicals.
TABLE 9 Commercial sources of chemicals
| Chemical | Manufacturers |
| BSA | Sigma No. A6793 |
| PVP 360 | Sigma No. P5288 |
| Ficoll 400 | Sigma No. F4377 |
| Salmon sperm DNA | Sigma No. D1626 |
| DTT | Sigma No. D9779 |
| Dextran sulphate | Sigma No. D8906 |
| SDS | Sigma No. L4390 |
| Glycin | Sigma No. G4392 |
| Deionized formamide | BRL No.540-5515UB |
| EDTA (TITRIPLEX 111) | Merck No. 8418 |
| DIECA | Merck No. 6689 |
| Phenol (analar grade) | Merck No.10188 |
| DNA detection system | BRL No.530-8239SA |
| Rio- 11 dUTP | BRL No.520-9507SA |
| Nick translation kit | BRL No.530-8160SB |
| Triton X100 | Merck No.11 869 |
| Chloroform | Merck No.2442 |
| Pentanol (isoamyl alcohol) | Merck No.979 |
| Calf thymus DNA | Sigma No, D8899 |
Sigma Chemical Company Inc,, PO Box 14508, St Louis, MO 63 178. USA; BRL, Gibco BRL France, 14 rue des Osiers, BP 7050,95051 Cergy Pontoise Cedex, France; Merck Schuchardt & Co., Eduard Buchner Strasse, D-8011 Hohenbrunn, Germany.
EXPERIMENTAL SET-UP
Schematically the technique can be divided into five steps:
EXPERIMENTAL PROTOCOL
Probe labelling
DNA probes. Purified recombinant plasmid DNA is labelled (by incorporation of either 32p sCTP, biotinylated dUTP or dCTP) by the technique of nick translation, using one of the several commercially available kits (e.g. BRL, Amersham).
RNA probes. After linearization of the purified recombinant plasmid downstream of the viral cDNA with a suitable restriction endonuclease, labelled RNA is produced by in vitro transcription using one of several commercially available kits (e.g. Promega, Biotec, Boehringer). Either 32p or biotin-labelled CTP is usually incorporated.
Sample preparation
Many different plant samples can be used, consisting of leaves, stems, tubers, barks or fruits (Figure 320). There is no standard protocol; each protocol should be optimized for a given host/virus combination. We present here a technique for the detection of plum pox virus, with additional advice on detection of other pathogens when appropriate.
Sample grinding One gram of plant sample is ground in 4 ml of grinding buffer using a pestle and mortar (or other apparatus such as an electric press or Polytron homogenizer when available), (Figures 321 to 323). It is extremely important to use a buffer that will optimize the signal-tonoise ratio. The extract is then clarified by centrifugation for 10 minutes at 10 000 rpm (Figure 324). The samples can, if necessary, be deproteinized by including one volume of a 1:1 mixture of water-saturated phenol and chloroform during the grinding. This step is optional for the use of radioactive probes but necessary when using biotinylated probes.
Sample denaturation. The nucleic acids contained in the supernatant are then denatured if necessary, to ensure good binding of the nitrocellulose and availability of the sequences for hybridization (Figure 325). This step is important for the detection of viroids but of no utility for most viruses. In a small microcentrifuge tube, 50 ?l of sample are added to 50 ?l of formaldehyde denaturation buffer. The mixture is then incubated for 60 minutes at 60°C (the length of this incubation should be reduced for viruses). At this point, samples are ready for spotting on the membrane. They can also be stored for up to several months at -20°C. We have found that concentration of the nucleic acids present in the extract by ethanol precipitation is detrimental since it usually increases the non-specific background reactions; it is therefore not recommended.
Nitrocellulose membrane preparation. Soaking of the membrane in a high-salt solution is required for proper binding of nucleic acids in the samples. The membrane is first soaked for 2 minutes in pure distilled water and then equilibrated for 10 minutes in 20X SSC buffer (Figure 326).
Sample application and fixation. Next, 20 ?l of sample are applied to the nitrocellulose membrane using a BRL "Hybri-dot" apparatus (Figures 327 to 330). Alternatively, 3 to 5 ?l of sample can be applied directly (using a micropipette) to nitrocellulose that has been airdried after soaking in 20X SSC. The membrane is then dried at room temperature (Figure 331) and baked for a further 2 hours at 80°C under vacuum to ensure stable binding of the nucleic acids to the nitrocellulose membrane (Figures 332 and 333). This can conveniently be achieved by using an electrophoresis slab gel drier or a vacuum oven. At this point, the membranes can be directly processed or sealed in a plastic bag (Figures 334 and 335) and stored (at 4°C or 20°C) for up to several months.
Hybridization reaction
Pre-hybridization. In order to prevent nonspecific binding of the probe to the membranes, they are pre-incubated in the hybridization mixture (pre-hybridization). The membranes are sealed in a plastic bag in the presence of 1 ml of hybridization buffer for each 10 cm² of membrane, taking care to avoid trapping any air bubbles (Figure 336). The bag is then incubated for 2 to 4 hours in a water bath at 42°C (Figure 337).
Probe denaturation. This step is included to remove any secondary structure of the probe and is especially important for DNA probes which are essentially double-stranded after the labelling reaction. A suitable quantity of probe is placed in a small disposable tube and incubated for 10 minutes (DNA probe) or 3 minutes (RNA probe) at 100°C in a bath of boiling water (Figure 338) and then quickly chilled by placing the tube in an ice-bucket.
Hybridization. The pre-hybridization buffer is discarded and replaced by the hybridization buffer to which the denatured probe has been added. Use approximately 1 ml of buffer containing radioactive probe of 1 to 2 x 106 cpm per ml or 200 mg per ml of biotinylated probe per 15 cm² of membrane (See techniques for determination of probe-specific activity, below). The plastic bag is then resealed and incubated in a water-bath overnight at 50°C (Figures 339 and 340).
Washing
After hybridization is completed, the membrane is removed from the plastic bag and washed in a small plastic tray (Figure 341). After washing, the nitrocellulose membranes should be airdried at room temperature (Figure 342).
DNA probes. Wash at room temperature for 5 minutes in three changes of washing buffer A, then proceed with two 15-minutes washes at 50°C in washing buffer B.
RNA probes. Carry out four 20-minute washes at 60°C in washing buffer C.
Hybrid detection
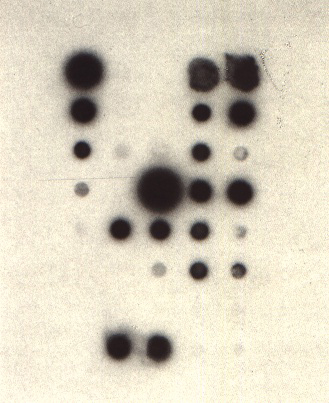
Radioactive probes. An X-ray film (Kodak XAR or equivalent) is exposed to the membrane for 24 hours at -70°C using intensifying screens (Figures 343 and 344). After autoradiography, the film is developed using Kodak LX 24 developer and Ilford Hypam fixer (Figure 345). Within the limits of linearity of the response of the film, the intensity of the spots is proportional to the concentration of viral RNA present on the membrane. No non-specific signal should be obtained with healthy plant controls.
Biotinylated probes. Several commercially available kits can be used for the detection of biotinylated probes (e.g. BRL). The composition of the buffers is given above.
The membranes are first soaked for 1 minute at room temperature in buffer 1, then for 20 minutes at 42°C in buffer 2 to saturate the protein-fixing sites on the membrane. They are then dried and baked for 10 to 20 minutes at 80°C under vacuum.
Following the treatment, the membranes are rehydrated for 10 minutes in buffer 2 and then incubated on a Petri dish in the streptavidin solution: 6 ?l of a 1-mg per ml streptavidin solution diluted in 3 ml of buffer 1. Incubate for 10 minutes at room temperature, shaking occasionally.
The membranes are then washed well with at least three changes of buffer for 3 minutes each time. Incubate on a Petri dish with 3 ml of buffer 1 containing 3 ?l of a solution of biotinylated polymers of alkaline phosphatase (polyAP) at 1 mg per ml. Incubate for 10 minutes at room temperature with occasional shaking.
Wash abundantly with two changes of buffer 1 and then with two changes of buffer 3. The developing solution should be prepared at the last moment in the following way: add 33 ?l of the nitro-blue tetrazolium solution to 7.5 ml of buffer 3. Mix thoroughly, then add 25 ?l of the 5-bromo-4-chloro-3-indolyl phosphate (BCIP) solution mix. Incubate the membrane in this solution in a sealed plastic bag protected from light.
Maximum colour development is usually achieved within 4 hours. To stop the development, simply wash the membrane in 20 mM Tris-HCI pH 7.5, 5 mM EDTA. The dried membranes can then be stored for several months in the dark to preserve the colour.
DETERMINATION OF THE PROBESPECIFIC ACTIVITY
Following the labelling reaction, the radioactive DNA probe ( 1 ?g is precipitated with ethanol, freed from the unincorporated labelled nucleotides by several 70 percent ethanol washes, dried and finally taken up in 100 ?l of sterilized distilled water. Then 2 ?l of the probe are mixed with 3 ml of a 10 percent trichloroacetic acid (TCA) solution along with 10 ?l of 3 mg per ml calf thymus DNA used as a carrier. The mixture is left for 30 minutes at 0°C and then filtered through a Whatman GF/C fibreglass filter. The filter is rinsed with 20 ml of a 5 percent TCA solution and then with 5 ml of ethanol before being dried. The radioactivity retained on the filter is then determined by liquid scintillation counting. The specific activity, in cpm per ?g, is given by cpm x 100/2.
Besides determining the specific activity of the probe, this technique can also help to calculate how much of the probe should be added to the hybridization reaction. Radioactive RNA probes can be counted in the same way.
For biotinylated probes, the result of the labelling reaction can be estimated by spotting dilutions of the probe on a membrane and comparing with a standard of known activity provided in the labelling kit.
FIGURE 319 Schematic representation of the system used for the detection of biotinylated probes
FIGURE 320 Plant sample consisting of leaves, bark, roots, tubers, fruits etc.
FIGURE 321 Plant sap is extracted using an electric press
FIGURE 322 A drop of sap is added to a grinding buffer contained in the microcentrifuge tube
FIGURE 323 Sap and grinding buffer are mixed using a vortex
FIGURE 329 Next, 20 ml of extract are spotted on the membrane while gentle vacuum is applied - A
FIGURE 330 Next, 20 ml of extract are spotted on the membrane while gentle vacuum is applied - B
FIGURE 331 The membrane is taken out of the blotting apparatus and air-dried
FIGURE 334 after drying, the membrane is placed in a plastic bag which is sealed on three sides - A
FIGURE 335 after drying, the membrane is placed in a plastic bag which is sealed on three sides - B
FIGURE 336 The pre-hybridization buffer is added to the plastic bag and the bag is completely sealed
FIGURE 338 The probe is denatured for a few minutes in a bath of boiling water
FIGURE 342 after washing, the membrane is air-dried
FIGURE 343 An X-ray film is exposed to the membrane - A
FIGURE 344 An X-ray film is exposed to the membrane - B
FIGURE 345 after autoradiography is completed, the X-ray film is developed and fixed
Extraction of closteroviruses from grapevine tissues
V. Savino
Closteroviruses are the cause of, or are associated with, diseases of grapevines such as leafroll and rugose wood. Except for grapevine closteroviruses A and B (GVA and GVB), which can occasionally be transmitted by inoculation of sap to herbaceous hosts, none of the other closteroviruses described to date are mechanically transmissible. Their isolation relies on extraction from naturally infected grapevine tissues.
PROCEDURE
The following procedure gives reproducible results.
1. Collect samples (10 g each) consisting of adult symptomatic leaves or cortex scrapings from mature dormant canes. Leaves may be placed in a freezer (-20°C) for 24 hours or longer before processing. Cortex shavings are recommended for American rootstocks, especially for pure Vitis rupestris and its hybrids, because extractions from leaves are negative.
2. Grind with pestle and mortar in liquid nitrogen until tissues are pulverized.
3. Add 5 volumes of the following extraction
buffer:
0.5 M Tris-HCI, pH 8.2
4. Stir in the cold (4°C) for 15 minutes.
5. Centrifuge at 5 000 rpm for 20 minutes.
6. Discard pellets. Save and filter supernatant liquid.
7. Centrifuge at 27 000 rpm for 2 hours.
8. Discard supernatant liquid. Resuspend pellets in a few drops of 0.02 M Tris-HCI buffer, pH 8.2, containing 0.01 M magnesium chloride.
9. Place a drop of the suspension on a dental wax bar or hydrophobic filter paper.
10. Float a freshly carbon-coated electron microscope grid on the drop for 15 minutes.
11. Stain with uranyl acetate and observe under the electron microscope.
Extraction of phloem-limited isometric viruses from grapevine tissues
G.P. Martelli
Phloem-limited viruses with isometric particles 25 to 30 nm in diameter are the cause of or are associated with diseases of the grapevine such as fleck, ajinashika disease and grapevine stunt. The latter two diseases have been reported only from Japan. None of these viruses are transmissible by inoculation of sap to herbaceous hosts. Their isolation relies upon extraction from naturally infected grapevine tissues.
PROCEDURE
A procedure that yields reproducible results for the recovery of the virus associated with fleck is the following:
1. Collect samples (10 g each) consisting of main veins and petioles or young succulent roots.
2. Grind with quartz sand in a mortar in the presence of 1 or 2 volumes of 0.05 M phosphate buffer pH 7.2 containing 5 mM 2 mercaptoethanol, 5 mM DIECA, 5 mM EDTA and 5 g per litre polyethylene glycol 6 000 (PEG).
3. Add 4 volumes of buffer to homogenate plus pectinase ( 10 g per litre) and cellulase (20 g per litre) and incubate at room temperature in the dark for 12 to 14 hours.
4. Strain through cheesecloth and centrifuge at 6 000g for 20 minutes.
5. Collect supernatant fluid, add 10 percent PEG and 1 percent NaCI while stirring, and incubate for 2 hours at 4°C.
6. Centrifuge at 10 000g for 20 minutes.
7. Discard supernatant and resuspend pellets in 0.05 M phosphate buffer, pH 6.8, containing 5 mM 2-mercaptoethanol, 5 mM EDTA and 0.2 M NaCI.
8. Stain with uranyl acetate and observe under the electron microscope.
Isolation and culture of Xylella fastidiosa
D.A. Golino
PROCEDURE
1. Prepare culture medium (PD3 according to Davis, Whitcomb and Gillaspie, 1981 ) as follows (quantities per litre):
Pancreatic digest of casein (tryptone, Difco), 4 g
Papaic digest of soy meal (Soytone, Difco or Phytone peptone, BBL), 2 g
Trisodium citrate, 1 g
Disodium succinate, 1 g
Hemin chloride stock (0.1 percent hemin chloride dissolved in 0.05 N NaOH), 10 ml MgSo4-7H2O, 1 g
K2HPO4, 1 5 g
KH2PO4, 1 g
Soluble potato starch, 2 g
Bacteriological agar (Difco), 15 g
Dissolve, autoclave and pour an appropriate amount into sterile Petri dishes.
2. Collect petioles from symptomatic grapevine leaves.
3. Cut petioles into fragments 2 to 3 cm long.
4. Surface sterilize by immersing for 5 to 10 minutes in 0.5 percent water solution of sodium hypochloride.
5. Rinse in three changes of sterilized distilled water under a sterile hood.
6. Express sap from petiole fragments by squeezing with sterile pliers or forceps and blot sap droplets directly on to culture medium. Alternatively, chop tissues finely with a razorblade in a sterile Petri dish or grind tissues in a sterile mortar in the presence of t .2 ml of sterile distilled water. Streak the slurry on to culture medium with a loop.
7. Incubate at 28°C under aerobic conditions.
REFERENCE
Davis, M.J., Whitcomb, R.F. & Gillaspie, A.G.M. 1981. Fastidious bacteria of plants and insects (including so-called rickettsia-like bacteria). In M.P. Starr, H.O. Stolp, H.G. Truper, A. Balows & H.G. Schlegel, eds. The prokaryotes: a handbook on habitats, isolation and identification of bacteria, p. 2171-2188. Berlin, Springer-erlag.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}