![]()
![]()
![]()
As with other vertebrates, also in fishes four haploid sperm cells develop from each of the primary spermatocytes, while only one haploid egg containing the second polar body develops from each of the primary oocytes. During the first meiotic division, crossing-over takes place which increases the genetic variability. In the case of normal fertilization the sperm penetrates the egg and the second polar body is excluded resulting in diploid progeny. For fast increasing at the in-breeding level, the gynogenesis technique was derived. In the process of gynogenesis the genetic material of the sperm is destroyed by radiation (UV, Cobalt 60, X-ray, etc.) and such sperms are used for fertilization. Since for normal development of the embryos the restoration of the diploid condition is required, a shock (cold, heat, chemical, etc.) is produced on the fertilized eggs.
If the shock is applied a few minutes after fertilization the diploid condition is restored by the retention of the second polar body (Appendix 1) while if the shock comes at the time of the first cleavage the diploid condition is restored by endomitosis (Appendix 2)
If the genetic material of the sperm is not destroyed these shocks will result in the development of triploid or tetraploid progeny (Appendix 3). The restoration of the diploid condition either by the retention of the second polar body or endomitosis has different consequences. Diploid condition restored by endomitosis normally results in 100 percent homozygous individuals (Appendix 2).
If the diploidy of zygotes is restored by inhibiting the second meiotic division (retention of the second polar body) heterozygous progeny will also develop. Thus, diploid gynogenetic progeny has a set of chromosomes derived from sister chromatids of half of a meiotic tetrad, heterozygous progeny will develop from a zygote containing sister chromatids with an odd number of exchanges between the centromere and the particular locus. The frequency of heterozygous progeny (r) is a direct measure of the recombination frequency - in this case the gene centromere distances, if F heterozygous female was used for gynogenesis. It has been pointed out by Nagy and Csanyi (1982) that the probability for a gene of recombination frequency r to be heterozygous in the ith gynogenetic generation is ri, while the coefficient of inbreeding is
Fi = 1-ri
The degree of uniformity or the degree of genotypic identity - the probability that two individuals selected from the ith gynogenetic generation are of the same genotype - either homozygous or heterozygous - with respect to the gene in question is:
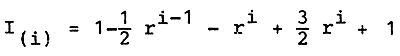
Distances between market loci and centromeres can be calculated according to the mapping functions derived for tetrad analyses (Barrat, 1954).
Therefore it is proposed to study the genetic markers by electrophoresis to obtain data for the above investigations.
The following eight different chromosome preparation techniques were experimented:
Chromosomes from blastodisc; aceto-orcein stained and fixed and squashed
Chromosomes from embryos; aceto-orcein stained, fixed and squashed
Chromosomes from hatchlings; aceto-orcein stained, fixed and squashed.
Chromosomes from embryos; alcohol-acetic acid fixed and air-dried
Chromosomes from hatchlings: aceto-alcohol fixed and air-dried
Chromosomes from rohu kidney: aceto-alcohol fixed and air-dried
Chromosomes from rohu gill epithelium: aceto-alcohol fixed and air-dried
Chromosomes from lucocyte of rohu
As the FARTC scientists are already familiar with the routine chromosome preparation technique from fish kidney, work was concentrated on adapting those methods (Nos. 1,2 and 3) which help to show the recombination of chromosomes immediately after the genetic treatment (e.g., induced polyploidy, intergeneric hybridization, etc.) and/or highly sophisticated methods to produce a good quality of spreads for chromosome banding or electron microscopic study (No.8).
The techniques adapted for the different chromosome preparations are as follows:
No. 1
Nos. 2 and 3
Nos. 4 and 5
Nos. 6 and 7
No. 8
Chromosome preparation from tissue culture.
Usually the best quality of metaphase chromosomes can be prepared
from tissue culture. For such culture, embryos, kidneys, testes
and leucocytes are commonly used. Chromosomes obtained from
leucocytes are of superior quality and can be used for either
banding or electron microscopic studies.
During the time of the consultancy, an experiment was carried out to adapt a method for rohu (Labeo rohita) leucocyte culture to determine optimal leucocyte density, culturing temperature and duration. As a result of the experiment the following methods are suggested:
Materials and equipment required:
Procedure
According to the results of this experiment, a higher number of leucocytes than suggested above (1 × 10 /ml) is not necessary. It is useful if the cultures are agitated every 24 hours to saturate the atmosphere of the culture bottle with pure oxygen so as to increase the cell division.
The advantages of the aceto-orecin stained squash techniques are that they are quick and simple, so that the chromosome aberrations, recombination problems, mortality during embriogenesis due to aneuploidy, etc., can be tested easily. However, the major disadvantages of this technique are that a large number of cells are broken during the preparation process, and/or the chromosomes are often in different focal planes. However, because of the advantages, it is proposed for use in the determination of ploidy degree, while the traditional kidney preparation technique for karyotype analyses and the leucocyte culture be used for electron microscopic study.
The first demonstration of differential staining of chromosomes was made possible by quinacrine fluorescence analyses. Later the Giemsa banding techniques became available. Then the use of proteolitic enzymes for demonstrating banding patterns in chromosomes was introduced. This last technique was modified by several authors and recently other techniques have been evolved (i.e., R banding, C banding, Potassium permanganate banding, etc.). With these banding methods, characteristic banding patterns can be obtained which permit the study of the chromatids. These methods facilitate the identification of homologous pairs and the secondary construction of the chromosomes also becomes visible.
Only three following chromosome banding techniques are listed because the proteolitic digestion C band techniques is well known to the counterpart scientists.
Controlled heating denaturation R-band technique
Following routine cell culture and chromosome preparation, slides are left for a few days. Then they are treated in phosphate buffer (Sorensen) or in Earle's Medium pH 6.5 for 10–20 min at 87°C, rinsed with tap water and stained with Giemsa.
Using this technique bands the reverse of the QM bands can be produced; and all chromosome pairs are clearly identifiable. The centromeric zones and secondary constructions are not stained. This technique has a great advantage in that slides previously stained by an ordinary technique may be treated directly without removing the stain, so that standard staining and banding denaturation of the same metaphase can be obtained.
Fluorescence QM banding technique
Conventionally alcohol acetic acid fixed and air-dried chromosome preparations are rehydrated in alcohol series and distilled water, then soaked in Sorensen's phosphate buffer 7.0 and stained in quinacrine mustard solution (50 ug/ml) for 20 min.
The chromosomes can then be investigated or photographed in a fluorescence microscope.
The QM is the best fluorochrome for chromosome identification. The bands are distinct, reproducible and the banding can be easily performed. As the Institute has a fluorescence microscope, this technique is obviously appropriate.
Giemsa banding techniques
Alcohol acetic acid fixed, air-dried chromosome preparations obtained from leuocyte culture are treated with NaOH (from 0.002, 0.007–0.07 N) for 30–120 sec (the optimal concentration and time have to be tested). After washing in alcohol and drying, the slides are incubated for about 12–18 hours in Sorensen's buffer at pH 6.8 (M/15 KH2PO4 × Na2HPO4) at 59°C.
Incubation without NaOH treatment can also produce good results. Staining is performed routinely in Giemsa.
With this Giemsa banding technique, a characteristic banding pattern can be obtained in fishes. Although the appearance of the chromosomes differs somewhat, this technique gives the same banding pattern as the trypsin method. The secondary constriction in the chromosomes are usually strongly stained. During the author's consultancy, R band technique and Giemsa banding technique were demonstrated.
Many studies were made on evolution of the fish karyotype and the cytotaxonomy of fishes. In addition, much research has recently been carried out to discover the relationships between species within a family.
Changes in chromosome number
The chromosome complements of fishes are very flexible. It has already been stated by cytologists that primitive, less specialized fishes have a higher number of chromosomes and more acrocentric chromosomes, while the more developed species have fewer and more metacentric chromosomes. It is also proved that Robertsonian evolution is a widespread phenomenon among fishes. Aneuploidy and polyploidy also often occurs among fishes. To date, hundreds of natural and induced polyploid fish are known, but many of them are not viable. Generally, if the fish has an odd number of chromosome sets, haploid, pentaploid, etc., they are not viable. The triploid fishes are exceptions. They are viable but sterile. In some cases the polyploidy is very useful, e.g, the common carp (Cyprinus carpio), which is a naturally tetraploid species. Generally the tetraploids are viable and reproductive, while hexaploids are viable but sterile. In fishes, chromosomal polyploid occurs (i.e., common carp, trout, etc.), possibly the results of translocations. The centric fusion of chromosomes is also known. In addition to these major changes, minor changes, like deletion and/or duplication of a specific locus or loci by unequal somatic sister chromatid cross-overs, also occur. Unequal meiotic division will also result in minor chromosome changes. These changes can be identified by karyotype analyses (major changes) or by studying the fine structure of the homologous chromosome pairs (minor changes).
The details of chromosomal sample preparations for electron microscopy were discussed and demonstrated. In section 2.2, a method is suggested for the chromosome preparation from leucocyte culture for electron microscopic observations.
After the culture is terminated, the cells should be harvested by centrifuging the suspension at 800 rpm for 10 min. The supernatant should be decanted and the cell suspension washed in physiological sodium chloride solution, then centrifuged again. After removing all except 1.5 ml of the supernatant, resuspend the solution. Remove all agglomerates because they are not suitable for electron microscopic studies. Place a drop of such a cell suspension on the top of D water and let the cells swell up by osmosis. Extract the cell suspension by a film-coated grid, and after drying, the chromosomes can be observed in electron microscope.
By electron microscopic observations, the structure and composition of fish chromosomes become apparent; the chromatids and chromatine fibres and their structural form can clearly be detected and the chromosomes in transition from interphase to metaphase can also be characterized.
It is known that ploidy causes an increase in size of blood cells. The erythrocytic size of polyploid fishes was investigated by several authors and they have found that the size of erythrocytes and their nuclei were larger in the case of various polyploid fishes than those of diploids.
During the consultancy a fast method was demonstrated to prove a triploidy by simple blood cell analysis instead of the difficult and labour-consuming karyological analyses. Heparinized blood was collected from the caudal vein. To determine the cell sizes, blood smears were prepared by the conventional method and stained with May-Grunwald solution. Slides were examined at a thousand-fold magnification. Major and minor axes of cell and nuclei were measured from 5 rohu, common carp and their hybrids respectively. Then the mean values of major and minor axis, characteristic for each subject were calculated. The surface and volume of erythrocytes and their nuclei should be counted using the following functions.

Multiple discriminant analyses should be aplied based on the measured parameters of erythrocytes for better discrimination of ploidity degree (Krasznai, 1984) (Appendix 4).
Knowledge and use of genetic markers are absolutely essential in genetic studies, especially in cytogenetic investigations, chromosome mapping, selection experiments, induced polyploidy and exploring the genetic origin of fish evolution.
In the last twenty years the use of electrophoresis, especially the use of enzymes, became very popular in studying the genetic background of fish populations. Isozymes markers should provide a convenient tool to understand developmental genetics and monitor various genetic treatments.
Although starch gel electrophoresis is still commonly used in research, there are some advantages in acrylamide gel electrophoresis which made this medium and technique superior.
Advantages of Acrylamide
Technique for Acrylamide Gel Preparation
For the formation of acrylamide gel the following working solutions should be prepared:
| A solution | B solution | |||||
| IN HCL | 48.0 | ml | IN HCL | 48.0 | ml | |
| TRIS | 36.6 | g | TRIS | 5.98 | g | |
| TEMED | 0.23 | ml | TEMED | 0.46 | ml | |
| DM Water up to | 100.0 | ml | DM Water up to | 100.0 | ml | |
| (pH = 8.9) | (pH = 6.7) | |||||
| C solution | D solution | |||||
| Acrilamide | 28.0 | g | Acrilamide | 10.0 | g | |
| BIS | 0.735 | g | BIS | 2.5 | g | |
| DM Water up to | 100.0 | ml | DM Water up to | 100.0 | ml | |
| E solution | F solution | |||||
| Riboflavine | 4.0 | mg | Sacharose | 40.0 | mg | |
| DM Water up to | 100.0 | ml | DM Water up to | 100.0 | ml | |
| Bridge buffer solution | Indicator stain | |||||
| TRIS | 6.0 | g | Bromphenolblue | 0.001 | g | |
| Glicin | 28.8 | g | DM Water up to | 100.0 | ml | |
| DM Water up to | 1000.0 | ml | ||||
| (pH = 8.3) | ||||||
| Before use dilute with DM Water 1:10 | ||||||
| Staining solution | Fix solution | |||||
| Amidoblack 10B | 1.0 | g | 7 percent acetic acid | |||
| 7 percent acid up to | 100.0 | ml | ||||
| G solution | ||||||
| Ammonium persulphate | 0.14 | g | ||||
| DM Water up to | 100.0 | ml | ||||
The technique described by Ornstein-Davis (1962) is still the best for separation of non-specific proteins from blood serum and muscle.
Three different layer of gels should be used. First, the small pore size - so-called running gel should be prepared in the following way:
Small pore size gel
| 1 | vol. of solution A |
| 2 | vol. of solution C |
| 1 | vol. of distilled water (DW) |
| 4 | vol. of solution G |
This mixture should be de-aerated and poured into the tubes or onto glass plates to within about 1.5 cm of the top. The solution must be overlaid with water to allow for polymerizetion, after which the water is removed and the large pore size gel is poured to within 3 mm of the top.
Large pore size gel
| 1 | vol. solution B |
| 2 | vol. solution D |
| 1 | vol. solution E |
| 4 | vol. solution F |
| (pH = 6.7) |
In slab electrophoresis to make place for the sample a teflon comb should be immersed into the gel. After polymerization the comb is removed and the gel is ready for use.
The large pore size gel is necessary to concentrate the sample into a narrow starting zone.
The samples should be placed either in gel or in 40 percent D-glucose in its place in the large pore size gel.
For the separation of different types of isozymes from different tissues, different bridge and gel buffers should be used to obtain the best solution.
Power supply
Almost any type of the comercially available units is sufficient. A power supply variable from 0 to 500 V and capable of continuous delivery to 250 mA is appropriate. In general the power supply should be capable of producing 12 V/linear cm of gel and 4 mA current per sample.
The best power supply allows for independent adjustment of voltage and amperage.
During the first 20–30 min of the electrophoresis the current should not exceed the 2 mA/sample after which it should be increased to 4 mA/sample until the end of the electrophoresis.
Cooling
The gell must be cooled, as it tends to heat up because of the electric current. The heat will inactivate the enzymes. A cold-room, if available, is an excellent place for electrophoresis although the separate cooling of the gel (either with built-in apparatus in which to locate a refrigerator in which to place the equipment) is necessary.
Staining
After electrophoresis the gels should be stained in case of transfer or NSP with 1 percent amidoblack in 7 percent acetic acid for 10–15 min, in case of enzymes specific stains should be used.
To start the electrophoretic work in FARTC, the author proposes to search for the following isozymes as indicated below:
Esterase (EST)
Lactate dehydrogenaze (LDH)
Malate dehydrogenaze (MDH)
Alcohol dehydrogenaze (ADH)
The reagents and the stains will vary according to the enzyme to be studied. The solution must be buffered at a specific pH, which is optimal for the isozyme, to provide good enzyme activity. In addition, the staining solution has to contain the substrate and all the necessary cofactors and an electron transport carrier PMS (Phenazine-methosulfate) and NBT (nitroblue tetrazolum). As the triphosphopyridine nucleotide is formed, it reduces the phenazine which in turn reduces the NBT. The NBT upon reduction is converted to an insoluble formazan which is blue. This phenazine tetrazolium system can be used to indicate the site of any dehydrogenaze enzyme which generates TPNH or DPNH (diphosphopyridine nucleotide).
The size of esterase isozymes are stained by the coupling of α-napthol with a dizonium salt (Blue RR) after the α-napthol is liberated from α-napthol acetate or α-napthol prorionate by the esterase activity.
EST
Gel buffer
0.02 M boric acid
The pH is adjusted to 8.6 with 2N NaOH
Bridge buffer
0.3 M boric acid
0.03 M sodium chloride
the pH is adjusted to 8.0 with 2N NaOH
Staining mixture
0.006 M α-naphtol acetate
200 mg Blue RR - salt
0.08 M TRIS
in 100 ml staining solution
(pH = 7.0)
LDH
Gel buffer is a 1:10 dilution of the bridge buffer
Bridge buffer:
0.1 M TRIS
0.1 M malic acid
0.01 M EDTA
0.01 M mg CI2
The pH is adjusted to 7.6 with 4N NaOH
Staining mixture:
0.535 M sodium lactate
0.000376 M DPN
0.000 163 M PMS
0.00031 M NBT
0.025 M TRIS
(pH = 7.5)
ADH
Gel buffer:
0.005 M histidine
The pH is adjusted to 8.0 with 2N NaOH
Bridge buffer:
0.41 M sodium citrate
The pH is adjusted to 8.0 with 0.41 M citric acid
Staining mixture:
7.5 ml of 95 percent athylalcohol
0.001 M DPN
0.000 163 M PMS
0.00043 M NBT
0.05 M sodium phosphate
in 100 ml of solution
(pH = 7.0)
MDH
Fixation
The NSP stained slabs or discs should be de-stained and fixed in 7 percent of acetic acid
All the isozymes developed in different staining solutions should be fixed in a solution of 5:4:1 ratio of water: methanol:acetic acid
Storage of gels
After fixation the gels can be kept and stored either in fix solution or dried
Method for drying
When the gel is ready to be dried (after de-staining, fixing) immerse the gel in an aqueous solution of 1 percent glycerol, 10 percent acetic acid for 30–40 min
Immerse 2 sheets of porous cellophane and boil for 5 min in the following solution.
5 g Na2 CO3
1.86 g EDITA
Up to 100 ml DW
Rinse the cellophane in DW
Place the gel between the two sheets of cellophane
Put one sheet of moistened filter paper onto the slab drier, add gel and cover with Saran wrap
Turn the heater to 80–82°C and connect to vacuum
During the author's consultancy in FARTC the counterpart scientists were trained in basic electrophoretic techniques. Blood serum and muscle tissue samples of Indian major carps, common carp and silver carp and their hybrids were used for gel electrophoresis. Polymorphisms in blood serum as well as in muscle tissue have been found.
The counterpart scientists had the background knowledge on theory of selection work and estimation of heretability. However, they had difficulties in the problem of designing a suitable experimental model. The author considered the design of a proper experimental model necessary to estimate the component of genetic variance because the knowledge so gained will determine the priority between selection and hybridization programmes. Hence the following programme was fully described and discussed to avoid any difficulties during conduct of the experiments.
To reduce the standard error to a minimum it is always advisable to work with as large a number of families as possible. About 50 Sire-Dam Cross is sufficient to minimize the error. From a practical point of view 48 crosses are given below:
EXPERIMENTAL MODEL FOR FAMILY SELECTION AND ESTIMATION OF SIRE AND DAM COMPONENT OF HERITABILITY
| Sires | Dams | Dams | Dams |
| 1 | 1 | 2 | 3 |
| 2 | 4 | 5 | 6 |
| 3 | 7 | 8 | 9 |
| 4 | 10 | 11 | 12 |
| 5 | 13 | 14 | 15 |
| 6 | 16 | 17 | 18 |
| 7 | 19 | 20 | 21 |
| 8 | 22 | 23 | 24 |
| 9 | 25 | 26 | 27 |
| 10 | 28 | 29 | 30 |
| 11 | 31 | 32 | 33 |
| 12 | 34 | 35 | 36 |
| 13 | 37 | 38 | 39 |
| 14 | 40 | 41 | 42 |
| 15 | 43 | 44 | 45 |
| 16 | 46 | 47 | 48 |
With such combinations the individuals of the cross are full sibs of each other while the individuals of the three subsequent crosses in the same row are half sibs of each other (like 1 ♂ × 1 ♀; 1 ♂ × 3 ♀). Using 16 Sires, 48 combinations can be produced. During the experiment these combinations have to be kept separate but the same environmental conditions have to be provided to all of them. Also the traits under investigation must be measured periodically and the data should be evaluated by the following model.
Yijk = μ + si + dij + eijk
Where Yijk = Corrected data of the kth individual from the population gained by mating the ith sire to the jth dam
μ = arithmetic mean
si = effect of ith sire
dij = effect of the jth dam on the ith sire
Estimation of the sire component of heritability
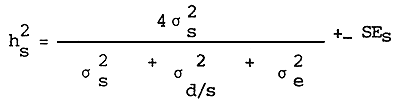
Estimation of the dam component of heritability
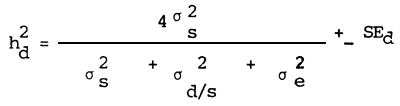
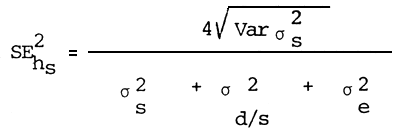
Estimation of the standard error of heritability
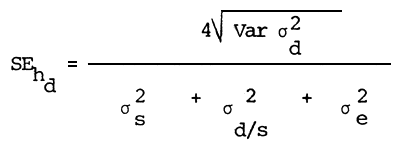
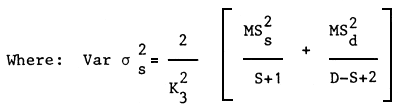
| Where: | MSs | = | Least square mean of sires |
| MSd | = | Least square mean of dams | |
| S | = | Number of sires | |
| D | = | Number of dams | |
| K3 | = | Mean number of offsprings from one Sire |
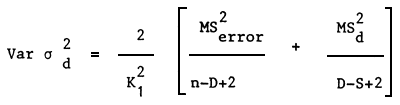
| Where: | MSError | = | Least square mean of sires |
| n | = | Total number of data | |
| K1 | = | Number of data per family |
Conclusions and recommendations.
The counterpart scientists of FARTC have acquired fairly good knowledge about the hybridization programme, gynogenesis and basic karyological studies.
During the consultancy the counterpart scientists were trained in chromosome preparation for light and electronmicroscopy, identification of genetic markers by gel-electrophoresis in studying cytogenetic evolution and crossing-over and in cell culture techniques.
Based upon the available facilities and the knowledge level of counterpart scientists, the following research projects are recommended for future priority programmes besides the existing ongoing projects:
Studies on karyology of Indian major carps, intergeneric hybrids, gynogenetic and polyploid populations.
Studies on genetic markers by electrophoresis for further studies of genetic background of fishes, chromosomal mapping, gene mapping, determination of gene-centromere distances and frequency of crossing over.
Electronmicroscopic studies on the ultrastructure of fish chromosomes.
Fish population genetics studies to determine the heritability of different traits and increasing the productivity of the existing strains/species.
![]()
![]()
![]()