![]()
![]()
![]()
E. HARMS
Universitaets-Kinderklinik
D-4400 Münster, FR Germany
Our present knowledge about the biogenesis and function of lysosomes would never have been established without the presentation of a variety of lysosomal disorders by nature's diversity. Research about lysosomes provides an excellent example of cooperation between medical and biological sciences. Medicine cannot make any progress without basic science, but the advancement of basic science also depends on clinical observations and questions. It has been estimated that 1 in 100 children has a monogenic disorder. Metabolic diseases amount to only about 10 to 20% of the monogenic disorders and almost exclusively follow an autosomal-recessive or X-linked inheritance.
About 250 inherited metabolic diseases are known today and more than 30 have been identified as lysosomal diseases, which thereby represent the largest organelle-specific group. About 70 metabolic diseases affect the central nervous system. Of the 250 diseases, only about 20% have a treatment protocol. With regard to the frequency of the particular disorders, we can expect 1 to 2 children in 1,000 to suffer from an inborn error metabolism.
Lysosomes degrade almost any type of physiological macromolecule (Table 1). A storage disease will result either from a missing degradation step or from defective release of the degradation product.
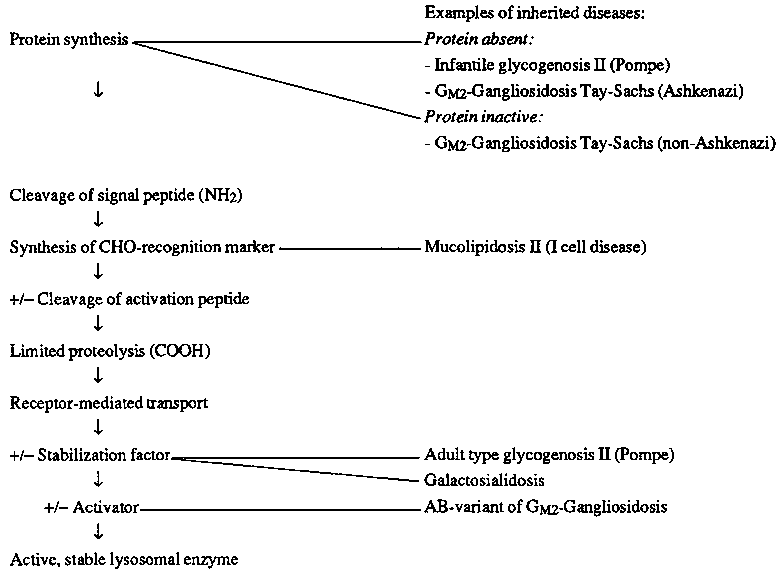
Lysosomal storage diseases can be classified into three groups, shown in Table 2. The most frequent by far is the absence of an enzyme needed for degradation. This lack of enzyme activity can be caused by mutations leading to absence of the protein or to defective enzyme protein, or by defects of activators needed for the degradation of some sphingolipids, or by defects of protective stabilizing proteins or peptide sequences. The largest group of lysosomal disorders are the deficiences of hydrolases involved in the degradation of heteroglycans, presenting as mucopolysaccharidoses, sphingolipidoses, glycoproteinoses and mucolipidosis I and IV.
The second type of lysosomal disease results from defective synthesis of the specific carbohydrate recognition marker as shown in mucolipidosis II and III. Research about these disorders has increased basic knowledge about the carbohydrate recognition marker.
The third group is likely to comprise transport defects through the lysosomal membrane. Examples in man are cystine storage in cystinosis and N-acetylneuraminic acid storage in Salla disease.
There still remain diseases which from morphologic observations are likely caused by a lysosomal dysfunction but for which the biochemical defect has not been established yet. The different forms of the large group of ceroidlipofuscinosis and the Chediak-Higashi syndrome are among these diseases.
The importance of genetic lysosomal diseases in the understanding of lysosomal biogenesis and function is outlined in Table 3. Steps in the formation of lysosomal enzymes, some of them facultative (+/-), are listed in this table and examples for genetic defects are given.
How can inborn errors of metabolism been treated? Table 4 summarizes the therapeutic strategies that have been used to treat inborn errors of metabolism. Only three treatment methods are of potential use for lysosomal diseases. Removal of toxic material is the rational for the treatment of cystinosis with cysteamine. When treatment is started early, the progress of the disease can be stopped. Cystinosis and Fabry's disease are diseases for which renal transplantation may be considered in the event of renal failure. Enzyme replacement might be a technique especially interesting for the treatment of lysosomal diseases, because the lysosomal enzymes exhibit a carbohydrate recognition marker for which receptors are expressed also on the cell surface pathway to the desired destination, the lysosome. However, practical realization of this concept is difficult, as summarized in Table 5.
Despite these difficulties, there have been many efforts to apply enzyme replacement for the treatment of human lysosomal diseases. All the earlier trials with discontinuous treatment have been unsatisfactory, because either the enzyme concentration was low, such as in plasma, or the side effects were not acceptable, such as after repeated buffy coat transfusions, or simply because of the technical problems to be overcome to gain the amount of enzyme needed for treatment. But the use of enzyme preparations for intravenous application is still being further developed. Molecular genetics allow the biosynthesis of large amounts of the missing enzymes. They can be modified to neoglycoproteins by adding carbohydrates, which can direct the enzymes to targeted organs. Another promising technique is to form more stable hydrolase-albumin complexes linked to antibodies that are directed to surface antigens of the targeted organ.
However, continuous enzyme replacement from transplanted organs seems to be more feasible. Bone marrow transplantation might be a viable treatment method for some lysosomal diseases. It has been shown that after bone marrow transplantation, a previously missing enzyme activity can be detected in organs such as the liver, spleen and kidneys. But for all diseases with CNS involvement, the blood-brain barrier seems to limit this approach. It is presently under discussion if this is completely true, because microglia cells, which comprise 5% of the brain cells, may be derived from bone marrow stem cells. If this proves correct, a very early bone marrow transplantation might even benefit those with lysosomal diseases with CNS involvement. However, in the diseases listed in Table 6, bone marrow transplantation already has been attempted as an experimental, sometimes desperate, trial. The presentation of data and the discussion of the results is very controversial and a definite recommendation cannot be given.
The strategy for the detection of a lysosomal storage disorder is outlined in Table 7. The cell fractionation technique we primarily use is free-flow electrophoresis. This technique enables us to purify lysosomes from almost any type of tissue culture cells, including lysosomal storage disorder cells with altered physical properties of lysosomes. It seems that free-flow electrophoresis is the method of choice for the isolation of lysosomes, especially from storage disease cells.
Many aspects of lysosomal diseases are still poorly understood (Table 8). There remain a great number of questions. One of these questions is: what makes the different phenoytypic expressions of the same enzyme defect? For instance, the same enzyme, alpha-iduronidase, is defective in two different diseases, the mucopolysaccharidoses type Hurler and Scheie-both with similar skeletal symptoms, but the first with severe brain damage, the second without any. It is most likely that the explanation of such differences will come from molecular genetics. An important question is why and how a lysosomal storage process causes a disease; this question will be answered by biochemists and biologists. These are related problems. The first concerns the specificity of a disease caused by a certain storage product. What we need is more insight into the biochemical sequelae damaging cells and organs and leading to a disease. The other unsolved questions are what happens to the stored material: what is its final destination, how does it get there, does it affect endocytosis, is it undergoing exocytosis, does it affect the function of other cells or organelles? These questions can be answered only by continuous research and collaboration among biochemists, cell biologists, geneticists and clinicians.
Table 1. Digestive function of lysosomes
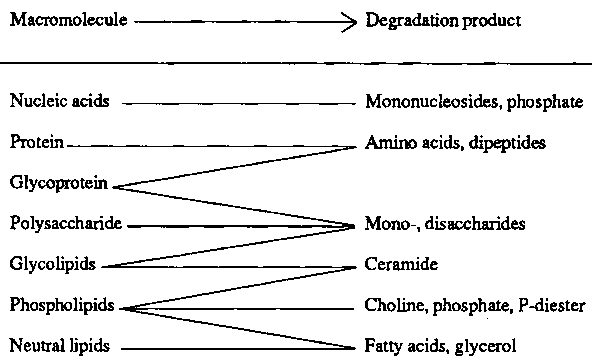
Table 2. Human lysosomal disorders
|
A: Defective enzyme activity | |
|
(missing or defective protein, defects of activator or protective protein) | |
|
|
- Mucopolysaccharidoses |
|
|
- Sphingolipidoses |
|
|
- Glycoproteinoses |
|
|
- Mucolipidoses I and IV |
|
|
- Glycogenosis type 2 |
|
|
- Acid lipase deficiency |
|
| |
|
B: Enzyme misplacement | |
|
|
- Mucolipidoses II and III |
|
| |
|
C: Transport defects | |
|
|
- Cystinosis |
|
|
- Sialic acid storage diseases |
|
| |
|
D: Unknown | |
|
|
- Ceroidlipofuscinoses |
|
|
- Chediak-Higashi syndrome |
Table 3. Biogenesis of lysosomal enzymes
Table 4. Therapeutic approaches to inborn errors of metabolism
|
1. |
Dietary restriction of substrate |
|
2. |
Replacement of missing product |
|
3. |
Removal of toxic storage material |
|
4. |
Activation of enzyme activity |
|
5. |
Replacement of missing enzyme |
|
6. |
Replacement of destroyed organ |
Table 5. Enzyme replacement for the treatment of lysosomal disorders - methods and difficulties
|
Methods | |
|
A: Discontinuous Therapy | |
|
|
1. Plasma infusion |
|
|
2. Leucocyte transfusion (buffy coat) |
|
|
3. Purified enzymes from urine, plasma, spleen and placenta |
|
B: Continuous Therapy | |
|
|
1. Organ transplantation |
|
|
2. Bone marrow transplantation |
|
Difficulties | |
|
- continuous supply of enzyme necessary | |
|
- ligand properties of enzyme versus receptor specificity of target organ | |
|
- one enzyme form may not reach all tissues affected | |
|
- blood-brain barrier? | |
Table 6. Bone marrow transplantation in human lysosomal diseases
|
Mucopolysaccharidoses |
Sphingolipidoses | |
|
MPS I |
(Hurler)* |
Metachromatic Leukodystrophy* |
|
MPS II |
(Hunter) |
M. Niemann-Pick* |
|
MPS IIIA, B |
(Sanfilippo) |
M. Krabbe* |
|
MPS IV |
(Morquio) |
M. Gaucher* |
|
MPS VI |
(Maroteaux-Lamy)* |
|
*Animal model available.
Table 7. Strategy for the detection of lysosomal storage diseases
|
Genetic transmission of a disease recognized (frequently associated with organomegally, +/- CNS damage) | |
|
| |
|
blood smear: lymphocyte vacuoles | |
|
| |
|
tissue biopsy: intracellular storage vacuoles (e.g., liver, kidney, nerve tissue, bone marrow) | |
|
|
|
|
Analysis of storage tissue: |
Identification of lysosomes: |
|
- histological differentiation by staining procedures |
- EM-cytochemistry |
|
- isolation and chemical analysis of storage material |
- immunocytochemistry |
|
|
- cell fractionation with co-purification of lysosomes/storage material |
Table 8. Open questions about lysosomal disorders
|
1. Different phenotypic expressions of the same defect (e.g., Huler/Scheie) | ||
|
| ||
|
2. Relation of storage product to clinical expression | ||
|
|
a) specificity of cell or organ alteration by a particular storage product (e.g., cystine, sialic acid) | |
|
|
b) the fate of undigested material: | |
|
|
|
- is the endocytic pathway affected? |
|
|
|
- is exocytosis involved? |
|
|
|
- is the cooperation of lysosomes with other cell compartments disturbed? |
![]()
![]()
![]()
{kind=link}