![]()
![]()
![]()
E. SHAW
The Friedrich Miescher-Institut
P O Box 2543, CH-4002 Basel, Switzerland
Use of protease specificity for inhibitor structure
Peptidyl fluoromethyl ketones
Peptidyl diazomethyl ketones
Peptidylmethyl sulphonium salts
Summary
References
The development of synthetic protease inhibitors that function by affinity-labelling offers the possibility of obtaining reagents that may be used with living systems to inactivate a target protease by covalent combination. The perturbation in the system should give some indication of the normal function of the protease. If the protease has a role in pathology, an effective inhibitor may have therapeutic value. The type of reagent we study is generally a low molecular weight peptide derivative. Irreversible combination with the target protease permits the inhibitor to resist displacement by normal substrates which are generally high affinity or present in relatively high concentration. In addition, the low molecular weight inhibitor may be made highly radioactive, permitting an analysis of the system with identification of the molecular target(s) and a correlation of the protein modification with some change in function. As the selectivity of the reagents improves, this type of application should become more common. It is already feasible with a number of proteases.
Our work has dealt with two of the proteinase classes, the serine and cysteinyl proteinases. (Metallo and aspartyl proteinases are insensitive to the types of reagents we study.) A knowledge of target enzyme specificity is essential for devising proteinase inhibitors that act by affinity labelling. Proteinases have an extended active centre that may combine with a substrate sequence of six or more amino acids. A familiar example is provided by the trypsin family of serine proteinases, which act on lysine and arginine residues. Individual members of this family are responsive to amino acid residues preceding the lysine or arginine residue. Thus, plasma kallikrein cleaves after a Phe-Arg-sequence in its natural substrate; factor Xa, after a Gly-Arg sequence; and thrombin, after Pro-Arg.
Chloromethyl ketones containing these sequences acquire a relative specificity for the corresponding protease (Table 1).1 Plasmin, in contrast, generally favours proteolysis at lysyl residues. Advantage may also be taken of differing responses to a D-amino acid residue in P3 or to various side-chain extensions that utilize an additional, possibly unique binding capacity in this region.2 In the case of serine proteinases, these reagents alkylate the active centre histidine residue.
However, peptidyl chloromethyl ketones also inactivate cysteinyl proteinases and for some in vivo applications may be too reactive. Nevertheless, it has been possible with D-Phe-Pro-ArgCH2Cl, a rapid inactivator of thrombin, selectively to inactivate this protease within the blood coagulation cascade by intravenous administration of the inhibitor.3
Some attention has been given to the development of analogous fluoromethyl ketones due to their diminished reactivity compared to chloromethyl ketones and the expectation that a proximity effect produced within the enzyme inhibitor complex would nevertheless promote covalent bond formation.4-7 This has essentially been demonstrated. Peptidyl fluoromethyl ketones are quite rapid in inactivating cysteinyl proteinases whose specificity they satisfy, yet are extremely inert to mercaptoethanol,6 a model for the abundant thiols found in biological materials. With respect to serine proteinases, the alkylation rate is slower than that of chloromethyl ketones, chiefly by 1-2 orders of magnitude (Table 2). This loss may well be acceptable if the gain is increased specificity in vivo. However, the fluoromethyl ketones are more difficult to obtain than the chloromethyl ketones.
A different type of a covalent bond forming group is the diazomethyl ketone. Peptides with this C-terminal function are unexpectedly stable. Serine proteinases are generally indifferent, whereas cysteine proteinases are typically very susceptible to members of this reagent class.
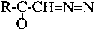
R = remainder of peptide without carboxyl
It is important at this point to comment on the specificity of cysteine proteinases known to us. Papain, the plant proteinase, is the best studied protease of this family, and, as an increasing number of mammalian proteinases are found to be homologous to it, a papain superfamily is identifiable, including the lysosomal cathepsins B, H, and L8 and the cytoplasmic calcium-activated neutral proteinases.9,10 There are other non-homologous cysteine proteinases of microbial origin.
Table 3. Specificity determinants in the papain family
|
P3- P2- P1 |
|
|
-Phe-X- |
Cathepsin L, B |
|
-Leu-Leu-X- |
Cathepsin L, Calpain |
Members of the papain family appear to bind substrates by positioning hydrophobic side chains in S2 and S3 of the active centre, therefore the nature of the side-chain of the residue whose peptide bond is being hydrolyzed is relatively unimportant. A phenylalanine residue at P2 has been shown to provide the basis of an effective series of reagents for inactivating cathepsin B and cathepsin L in which the P1 residue, X, may be basic, hydrophobic, small or large. Although the nature of the amino acid in P1 does not seem to be specificity determining, it was found that cathepsin L tolerates certain bulky side chains better than cathepsin B in this position and the exploration of this difference led to a considerable enhancement of selectivity. For example, Cbz-Phe-Tyr(O-t-Bu)CHN2 at 10-8 M inactivates cathepsin L with a t½ of 5.7 mins, a rate 2.5 × 104 greater than its action on cathepsin B.11
Cellular studies of the uptake and metabolic effect of peptidyl diazomethyl ketones have shown-either a drop in protein turnover in the case of cathepsin B and L inactivators,12,13 a drop in residual intracellular proteolytic activity14,15 or the restoration of an enzymatic activity (aryl sulphatase). The latter was observed in the case of a lysosomal storage disease involving an aberrant but still functional aryl sulphatase that was destroyed by catheptic proteolysis.16
Similar considerations of specificity apply to the calcium-activated neutral protease, calpain, found in two forms in the cytoplasm. This proteinase is attracted to hydrophobic amino acid side chains that bind in the S2 and S3 subsites of the substrate-binding region of the active-centre, as found by Sasaki et al.17 from examination of a variety of peptide substrates. A Leu- Leu- sequence is particularly favoured. This led to the synthesis of small peptide inactivators for calpain, such as Leu-Leu-TyrCH2Cl18 and Cbz-Leu-LeuMetCHN2,19 which rapidly inactivate calpain in vitro. Because cathepsin L also has affinity for similar residues in P2 and P3, these inhibitors also inactivate that protease.19 However, cathepsin L inactivators containing a phenylalanine in P2 have no effect on calpain; therefore, ambiguity can be resolved with the use of two inhibitors at the present time. It may be that further work will provide a specific calpain inactivator. It can be expected that such an inhibitor would have wide application in cellular studies on the role of this protease.
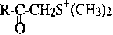
R = remainder of peptide without carboxyl
A third class of affinity-labelling inhibitors was modelled on S-adenosyl methionine as a biological alkylating agent.20 Peptidylmethyl sulphonium salts are particularly effective for inactivating cysteine proteinases and have the advantage of being accessible with amino acid side chains such as arginine whose incorporation into other inhibitor types is chemically difficult.21,22
In connection with protein trafficking, it may be of interest that inhibitors of this type have been applied to the study of prohormone processing. Rat proinsulin is converted to insulin by two endo-proteolytic steps and subsequent trimming. The activities have been separated from insulin secretory granules of rat insulinoma tissue by Hutton and his colleagues23 as Ca++-dependent proteases with a low pH-optimum. One of the proteolytic splits follows a Lys-Arg sequence and the other an Arg-Arg- sequence. The activity responsible for the post Arg-Arg cleavage is highly sensitive to inactivation by Ala-Arg-ArgCH2S+ (CH3)2, but considerably less (orders of magnitude) to Ala-Lys-ArgCH2S+- (CH3)2. On the other hand, this reagent is more effective in inactivating the enzyme cleaving after Lys-Arg, as expected. The nature of the proteases involved is not yet certain, although this sensitivity is characteristic of cysteine proteases perhaps other types have not yet been adequately examined.
We have described several classes of peptide derivatives that act as affinity labelling inactivators of serine and cysteine proteinases. Some of these have been shown to enter cells and inactivate the target protease as demonstrated by protein chemistry. In other cases cellular processes have been blocked but the protease involved has not yet been identified. Extension of these methods can be expected to shed further light on protein metabolism.
1. KETTNER, C. and E. SHAW. 1982. Meth. Enzymol. 80: 826-842.
2. GANU, V.S. and E. SHAW. 1987. Thrombosis Res. 45: 1-6.
3. COLLEN, D., O. MATSUO, J.M. STASSEN, C. KETTNER and E. SHAW. 1982. J. Lab. Clin. Med. 99: 76-83.
4. RASNICK, D. 1985. Anal. Biochem. 149: 461-465.
5. RAUBER, P., H. ANGLIKER, B. WALKER and E. SHAW. 1986. Biochem. J. 239: 633-640.
6. ANGLIKER, H., P. WIKSTROM, P. RAUBER and E. SHAW. 1987. Biochem. J. 241: 871-875.
7. ANGLIKER, H., P. WIKSTROM, P. RAUBER, S. STONE and E. SHAW. In press. Biochem. J.
8. TAKIO, K., T. TOWOTARI, N. KATANUMA, D.C. TELLER and K. TITANI. 1983. Proc. Natl. Acad. Sci USA 80: 3666-3670.
9. OHNO, S., Y. EMORI, S. IMAJOH, H. KAWASAWI, M. KISARAGI and S. SUZUKI. 1984. Nature 312: 566-570.
10. SHAW, E. and C. KETTNER. 1981. Acta Biol. Med. Germ. 40:1503-1511.
11. KIRSCHKE, H., P. WIKSTROM and E. SHAW. 1988. FEBS Lett. 228: 128-130.
12. SHAW, E. and R.T. DEAN. 1980. Biochem. J. 186: 385-390.
13. GRINDE, B. 1983. Biochem. Biophys. Acta 757: 15-20.
14. SUTHERLAND, J.H.R. and L.M. GREENBAUM. 1983. Biochem. Biophys. Res. Communs. 110: 332-338.
15. GREEN, G.D. and E. SHAW. 1983. Arch. Biochem. Biophys. 225: 331-337.
16. VON FIGURA K., F. STECKEL, J. CONARY, A. HASILIK and E. SHAW. 1986. Am. J. Hum. Genet. 39: 371-378.
17. SASAKI, T., T. KIKUCHI, N. YUMOTO, N. YOSHIMURA and T. MURACHI. 1984. J. Biol. Chem. 259: 12489-12494.
18. SASAKI, T., T. KIKUCHI, I. FUKUI and T. MURACHI. 1986. J. Biochem. 99: 173-179.
19. CRAWFORD, C., R.W. MASON, P. WIKSTROM and E. SHAW. In press. Biochem. J.
20. SHAW, E. 1988. J. Biol. Chem. 263: 2768-2772.
21. RAUBER, P., B. WALKER, S. STONE and E. SHAW. 1988. Biochem. J. 250: 871-876.
22. ZUMBRUNN, A., S. STONE and E. SHAW. In press. Biochem. J.
23. DAVIDSON, H.W., C.J. RHODES and J.C. HUTTON. 1988. Nature 333: 93-96.
Table 1. Inhibitor selectivity is a function of sequence (rates of inactivation 10-3 × kapp/[I] (M-l sec-1))
|
Inhibitor |
Plasma kallikrein |
Factor Xa |
Thrombin |
Plasmin |
|
Pro-Phe-ArgCH2Cl |
23.3 |
0.22 |
0.02 |
0.032 |
|
Ile-Glu-GIy-ArgCH2Cl |
4.8 |
32.0 |
0.5 |
0.075 |
|
D-Phe-Pro-ArgCH2Cl |
8.0 |
4.5 |
11500.0 |
0.67 |
|
D-Lys(Bz)-Phe-LysCH2Cl |
|
0.005 |
0.23 |
47.0 |
Table 2. Comparisons of peptidyl chloromethyl and fluoromethyl ketones
|
|
pH |
Ki (m M) |
ki (s1) |
Ref | |
|
Cathepsin B | |||||
|
|
Cbz-Phe-PheCH2Cl |
5.4 |
0.23 |
0.21 |
5 |
|
|
Cbz-Phe-PheCH2F |
5.4 |
0.14 |
0.055 |
|
|
Thrombin | |||||
|
|
D-Phe-Pro-ArgCH2Cl |
8.0 |
0.025 |
0.115 |
7 |
|
|
D-Phe-Pro-ArgCH2F |
7.0 |
0.25 |
0.0015 |
|
|
Plasmin | |||||
|
|
Ala-Phe-LysCH2Cl |
7.0 |
0.83 |
0.003 |
6 |
|
|
Ala-Phe-LysCH2F |
7.0 |
5.0 |
0.00042 |
|
![]()
![]()
![]()