![]()
![]()
![]()
R.B. KELLY, L. MATSUUCHI and T. BURGESS*
(* Current address: Department of Biological Sciences, University of California, Santa Barbara, California, USA.)
Department of Biochemistry and Biophysics
University of California
San Francisco, California, USA
Sorting of proteoglycans into secretory granules
Conclusion
References
Prior to secretion, secreted proteins are found in cytoplasmic secretory vesicles in eukaryotic cells. Secretion of protein almost universally involves fusion of the secretory vesicle to the plasma membrane. Although the mechanism of protein secretion appears to be the same in all eukaryotes, there is considerable variation in whether or not secretory vesicles accumulate in the cytoplasm, in how the secretion rate is regulated and where secretion occurs in the cell. Cells such as lymphocytes store very little assembled secretory protein in their cytoplasm, while cells such as exocrine cells and mammary epithelial cells (Table 1) store secreted proteins in large secretory vesicles that characteristically have electron dense cover.1,2 The rate of secretory vesicle fusion may be affected by no known stimulus, as in the case of lymphocytes or mammary epithelial cells, or it may be regulated by an intracellular second messenger, as is the case for endocrine, exocrine and neuronal cells, for example. Finally, the sites of secretory vesicle fusion may be restricted. As can be seen from Table 1, secretion can be apical, basolateral or non-polarized. Regulated secretion is found only in cells that have storage vesicles, consistent with a model in which regulated secretory cells store their newly synthesized secreted products until an appropriate extracellular signal triggers phasic release.
Newly synthesized membrane proteins reach the cell surface in transport vesicles. Because such transport vesicles have an internal volume, they will contain all types of newly synthesized secretory protein unless an exclusion mechanism operates. Conversely, storage secretory vesicle membranes will carry membrane proteins to the surface, unless there is a membrane protein exclusion mechanism. To test whether membrane proteins were excluded from secretory granules, we purified secretory granules from the mouse pituitary cell line, AtT-20, and showed that they lacked a newly synthesized membrane protein, gp70.3 We concluded that plasma membrane proteins were excluded from secretory granules and exited the cell by a different exocytotic pathway. This pathway we named the constitutive pathway, to distinguish it from the exocytotic pathway for release of storage granule contents, which had previously been called regulated secretion The constitutive pathway was defined initially as the pathway that plasma membrane proteins take to the surface of cells. Externalization of membrane proteins was found to be stimulus-independent, whereas externalization by the regulated pathway was stimulus-dependent. Since some basal secretion can take place from the regulated pathway even in the absence of a stimulus, constitutive secretion is not the same as stimulus-independent secretion in our usage. Others equate the two, and so include in constitutive secretion stimulus-independent release from secretory granules.4 We prefer to retain a definition of pathways that is based on partitioning newly synthesized proteins.
In the pituitary cell line, AtT-20, both membrane proteins and secreted proteins can be excluded from the secretory granule.5 The extracellular protein, laminin, appears to be one of the secreted proteins not in secretory granules.6 The simplest model that fits currently available data is that the secreted proteins that are excluded from the secretory granule exit the cell by the constitutive pathway taken by excluded membrane proteins. Direct proof of this model is lacking. There may be more than one pathway for constitutive secretion; fusion of immature granules could occur on the regulated pathway.4,7
We have explored how different proteins are transported to the cell surface by transfecting DNA encoding secretory proteins into secretory cells. We discovered that proteins fell into two categories, those transported like ACTH, by the regulated pathway, and those transported like laminin, by the constitutive pathway. The data for two such proteins - trypsinogen and the kappa light chains of immunoglobulin - are given in Table 2 and compared with the secretion of ACTH, monitored by radio-immunoassay. There is a slowly turning-over pool for both ACTH and trypsinogen, which can be diminished by stimulation. In contrast, there is no pool with an unusually long half-time for the kappa chains, nor is the size of the pool affected by stimulation. The secretion rate of trypsinogen and ACTH is increased four to six times by a stimulus, while kappa secretion rates are unaffected.
We have attempted to look at what features of trypsinogen cause it to enter the regulated pathway using in vitro mutagenesis. Data on three mutant trypsinogens are compared with a wild type in Table 3. The sorting index for the wild type calculated in this experiment is 0.10.8 One of the mutants, TB08, has approximately the same sorting index, while two (TB09 and TB10) appear to be sorted more effectively than the wild type. Increases of sorting efficiency on mutagenesis have also been reported for defective insulin.9
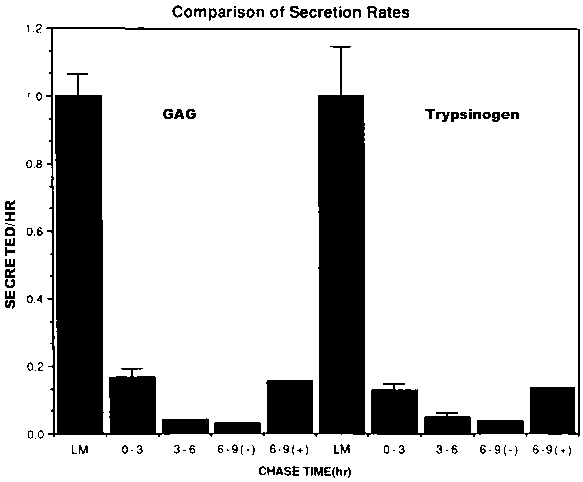
We have compared the sorting of a free oligosaccharide chain to protein sorting. Dense core secretory granules contain a sulphated proteoglycan of the chondroitin sulphate type.5,10 The sorting of free glycosaminoglycan (GAG) side chains, unattached to polypeptide chains, can be determined directly by labelling cells with 35S-sulphate in the presence of the GAG chain initiator, umbelliferyl xyloside.10 When we apply our conventional sorting assay to the partitioning of free GAG chains between the pathways, we get a surprising result (Figure 1), namely that the sorting of trypsinogen and GAG chains are indistinguishable. It is unlikely that a protein and a carbohydrate can be recognized by identical sorting machinery. We are considering the following alternative explanations. (1) The GAG chains are synthesized in the secretory vesicles after sorting has occurred. If so, the remarkable similarity between the sorting indices for proteins and GAG chains would then be fortuitous. (2) The sorting index represents relative volume flow from the Golgi region to the two pathways, and constitutively secreted proteins are excluded from the regulated pathway. (3) Proteins and GAG chains condense together into a dense core in immature granules prior to sorting and are sorted together.
In addition to GAG secretion, we have re-examined the sorting of an intravesicular chondroitin sulfate proteoglycan that we had characterized earlier.10 Measuring the sorting index of this proteoglycan by conventional techniques,8 we discovered that the sorting index was eight times higher than that of trypsinogen. Such apparently efficient sorting is strongly reminiscent of that of the hybrid protein created by deleting the C-peptide from proinsulin.9 There are at least two possible interpretations of these findings, proteoglycan could be very efficiently sorted, perhaps, for example, by forming the condensed core to which the hormones adsorb. Alternatively, the proteoglycan that we measure could be generated largely or exclusively within the secretory granule. This would give a misleading high sorting index. To incorporate the data on the hybrid insulin molecule sorting into these models we would propose either that the hybrid molecule, perhaps because of its inability to generate correct disulfide bonding, condenses on a core matrix more readily.9 An alternative explanation is that the antigenicity of the mutant proinsulin is generated only in the milieu of the secretory granule.
While there is no doubt some proteins are sorted into secretory granules and some are not, the mechanism of sorting remains obscure. It certainly does not seem to be as simple as targeting lysosomal enzymes to the lysosome. Mutations in proteins can increase their efficiency of sorting into pathways. Increases in efficiency would be consistent with models of sorting in which proteins need to come out of solution and condense to a solid matrix prior to sorting, a model consistent with current data from electron microscopy.11,12 The probability of coming out of solution to form a matrix could readily be enhanced by mutation of the three-dimensional structure. Likewise, a core model could explain the co-sorting of free GAG chains and proteins. Proteins excluded from the regulated pathway would be those excluded from the forming core, although the accuracy of current data would also fit with lack of condensation, but no exclusion from the core. Even if matrix formation underlies protein sorting in secretory cells, the problem remains how selected membrane proteins, the integral membrane proteins of the secretory granule, are segregated with the dense protein cores.
1. TARTAKOFF, A.M., P. VASSALLI and M. DETRAZ. 1978. J. Cell Biol. 79: 694-707.
2. PALADE, G. 1975. Science 189: 347-358.
3. GUMBINER, B. and R.B. KELLY. 1982. Cell 28: 51-59.
4. VON ZASTROW, M. and J.D. CASTLE. 1987. J. Cell Biol. 105: 2675-2684.
5. MOORE, H.P., B. GUMBINER and R.B. KELLY. 1983. J. Cell Biol. 97: 810-818.
6. BURGESS, T.L., C.S. CRAIK, L. MATSUUCHI and R.B. KELLY. 1987. J. Cell Biol. 105: 659-668.
7. RHODES, C.J. and P.A. HALBAN. 1987. J. Cell Biol. 105: 145.
8. MOORE, H.P.H. and R.B. KELLY. 1985. J. Cell Biol. 101: 1773-1781.
9. POWELL, S.K., L. ORCI, C.S. CRAIK and H.P.H. MOORE. 1988. J. Cell Biol. 106: 1843-1851.
10. BURGESS, T.L. and R.B. KELLY. 1984. J. Cell Biol. 99: 2223-2230.
11. KELLY, R.B. 1985. Science 230: 25-32.
12. TOOZE, J., S.A. TOOZE and S.D. FULLER. 1987. J. Cell Biol. 105: 1215-1226.
Table 1. Secretion patterns in eukaryotes
|
Tissue |
Storage in secretory vesicles |
Regulated |
Polarity |
|
Exocrine |
+ |
+ |
apical |
|
Mast cell |
+ |
+ |
- |
|
Endocrine |
+ |
+ |
basal (?) |
|
Mammary epithelia |
+ |
- (?) |
apical |
|
Lymphocytes |
- |
- |
- |
|
Liver |
- |
- |
basolateral |
Storage of secretory vesicles is positive when secretory vesicles usually with characteristic dense cores accumulate in the cytoplasm. Secretion is regulated when a known extracellular signal increases or decreases the rate of secretory vesicle fusion. Polarity of secretion is defined for epithelial cells. To what extent endocrine cells should be considered as polarized is not clear at this time.
Table 2. Comparison of secretion properties in AtT-20 cells
|
|
ACTH1 |
TRYPSINOGEN2 |
KAPPA2 |
|
Half-time of storage pool (h) |
18.0 ± 4.0 (3) |
14.8 ± 1.7 (4) |
1.8 ± 0.5 (3) |
|
Released on stimulation (%h) |
12.3 ± 1.3 (3) |
17.1 ± 1.0 (4) |
-1.3 ± 2.0 (3) |
|
Secretion rate (stimulated/rest) |
5.6 ± 1.5 (3) |
4.1 ± 1.5 (5) |
0.9 ± 0.5 (3) |
The releasable pool is considered to be the entire intracellular ACTH concentration measured by radioimmune assay, or the amount that remains in the cell extract after an overnight label with 35S-methionine, and a several-hour chase. The half-time is calculated from the rate of release from the pool per hour. The amount released on stimulation is the fraction of the pool released in one hour in the presence of a stimulus, less the fraction released in the absence of a stimulus. The ratio of the secretion rate in the presence of the stimulus, in this case, 8-Br-cAMP, to that in its absence is given. The figures in parentheses are the numbers of measurements.
1 Measurements made by radioimmune assay.
2 Measurements made after a 15-hr label with 35S-amino acids followed by a 3 - to 6-hr chase.
Table 3. Sorting efficiencies of trypsinogen mutants
|
Trypsinogen |
Wild type |
TB08 |
TB09 |
TB10 |
|
Sorting index |
- |
70 - 79 |
145 - 149 |
243 - 245 |
|
Region deleted |
0.10 |
0.08 |
0.20 |
0.21 |
The sorting indices were measured exactly as described by Moore & Kelly (1985) and Burgess et al. (1987).
Figure 1. Comparison of chase kinetics for glycosaminoglycans and trypsinogen. The data for trypsinogen were generated exactly as described earlier (Burgess et al., 1985) in that cells were labelled with 35S-amino acids for 15 hr. The rate of release at the end of the labelling period was determined by immunoprecipitation of labelled trypsinogen. Similarly, the amount secreted during chase periods of 0 to 3 hr. 3 to 6 hr and 6 to 9 hr. Also included is the increment in release rate between 6 and 9 hr, when 5 mM 8 Br-cAMP was included in the chase medium. To label free GAG, the GAG chain initiator, umbelliferyl xyloside, was added to the medium in the presence of 35S-sulphate, as described previously (Burgess and Kelly, 1984). The secreted GAG chains, identified by their characteristic "staircase" appearance on polyacrylamide gels, were quantified by elusion from the gels.
![]()
![]()
![]()